Life Extension Magazine®
CoQ10 fuels cellular respiration in the “energy factories” of the cell, called mitochondria. CoQ10 also protects cells and blood lipids from oxidative damage.
As science begins to unravel the mechanisms of life and death at the cellular level, ongoing research throws a spotlight onto the mitochondria. The mitochondria not only sustain life by generating energy, but end life by triggering programmed cell death (apoptosis). Indeed death and degeneration begin in the mitochondria as cellular respiration declines and “cellular suicide” increases.
This article examines how mitochondrial mutations, bioenergetic decline and oxidative stress can interact to bring about aging and degenerative disease. CoQ10—the mitochondrial agent par excellence—emerges as a bioenergetic/antioxidant therapy for the disease of aging. We then consider two major families of age-related disease, the metabolic syndrome (which gives rise to cardiovascular disease and diabetes) and cancer.
Bioenergetic aging
Aging reduces bioenergetic capacity and the ability to respond to stress—in a word, vitality declines, on the level of both the cell and of the organism. This brings about a progressive decline in function and susceptibility to age-related diseases.
A theory of aging that elegantly weaves together the most productive threads of aging research was proposed by Australian scientist Anthony Linnane in 1989. Linnane began by observing that mutations accumulate in mitochondrial DNA with age. Several years later he was able to show that less than 5% of the mitochondrial DNA from the muscle tissue of a 90 year old subject was intact.
Mitochondrial DNA defects increase exponentially in some body tissues after age 30 or 40. The tiny package of DNA in the mitochondria is especially vulnerable because it lacks some of the systems that protect and repair DNA in the cell nucleus. More defects have a chance to accumulate in the mitochondrial DNA of cells that cannot be replaced in the heart (cardiomyocytes), brain (neurons) and skeletal muscle (myocytes). We don't know exactly how these defects develop, but oxidative stress and errors in mitochondrial DNA replication appear to be primary causes. A recent study found that the rate of one well-known mitochondrial DNA defect (the “common deletion”) corresponds to the level of lipid peroxidation in the mitochondria and increases with age.
CoQ10 protected mitochondrial DNA in a study of mice given a drug that generates high levels of oxidative stress. The drug, doxorubicin, causes severe lipid peroxidation in mitochondria. In this experiment, a third of the mice given the drug without CoQ10 developed a mitochondrial DNA deletion. When CoQ10 was given along with the drug only 7% of the mice developed the deletion, and when the CoQ10 dosage was increased none of the mice developed the mitochondrial DNA deletion.
According to Linnane's theory, as defects accumulate in mitochondrial DNA, cellular energy production falls off (see Figure 1). In other words, it is the deterioration of mitochondrial DNA that drives bioenergetic decline as we age (see the sidebar “Mitochondrial Mutations: Cause of Aging?”). This is logical since mitochondrial DNA holds much of the genetic blueprint for the machinery of cellular respiration. When the blueprint is damaged, the cellular respiratory chain becomes defective. Many years later technology was developed to test this proposition, and again Linnane was proved correct. In 1998 he was able to demonstrate a close correlation between mitochondrial mutations and cellular energy production in individual human muscle fibers.
The random mutations in mitochondria create a “bioenergetic mosaic.” Cells in the same tissue produce different amounts of energy depending upon their degrees of mitochondrial damage. Some cells produce relatively little energy, some a moderate amount and some a large amount of energy. In a five year old's tissues we do not see this mosaic since energy production is high in nearly all cells. However a pronounced “mosaic effect” develops after about age 40. This occurs to a different extent in different tissues depending upon their rates of bioenergetic aging. Some tissues such as skeletal muscle appear to age very quickly from a bioenergetic viewpoint, while others such as cardiac muscle age at a moderate rate, and others such as liver tissue age quite slowly. This line of research also illustrates the fact that cellular aging proceeds at different rates in different people, since people of the same age often show markedly different bioenergy mosaics.
Linnane's theory of the “universality of bioenergetic disease” holds that this mutation-driven bioenergetic decline is a major factor in the degenerative diseases and general frailty of old age. Many lines of recent research converge on the mitochondria as centers of cellular aging. If this mitochondrial theory of aging is correct, the foundation of cellular vitality lies in the mitochondria.
Mitochondrial Mutations: Cause of Aging?
Skeptics have argued that not enough mitochondrial DNA is damaged to account for the declining cellular function seen in aging. Recently published research from Caltech examines the mitochondrial DNA in connective tissue cells (fibroblasts) from individuals of all ages, including stored cells taken many years apart. The study demonstrates high levels of age-related mutations in a pivotal portion of mitochondrial DNA—the portion that controls the replication (reproduction) of mitochondrial DNA—of some individuals. For example, one specific mutation in this genetic region was shared by the majority of individuals in the study over 65 years of age, but was absent in younger individuals. In addition, the study documents seven other mutations in elderly individuals at genetic spots critical for the replication of mitochondrial DNA.
Since mitochondrial DNA undergoes continuous turnover in both renewable and non-renewable cells, mutations in the genes controlling its replication could have far-reaching consequences. Broad follow-up research is needed to assess the impact of such genetic defects on mitochondrial function. It is reasonable to hypothesize that they could lead not only to a snowballing of mutations as the mitochondria replicate, but also to defects in the genetic blueprint for cellular respiration, and increased oxidative stress (due to increased oxidant generation during cellular respiration or compromised antioxidant defenses), which would in turn cause more oxidative damage to mitochondrial DNA and to the cellular respiratory chain. Such a vicious cycle is depicted in the sidebar “A Model of Bioenergetic Aging.”
These findings complement studies of DNA pointing to age-related disturbances in genes that control cell division (mitosis), which commonly leads to aberrant cell division and chromosome instability. More broadly, scientists hypothesize an age-related “mutator phenotype” that accelerates the accumulation of genetic mutations. In this view, DNA damage, chromosomal instability and chronic exposure to genotoxins such as oxidative stress feed back upon each other, contributing particularly to cancer development.
One of the proposed causes of the mutator phenotype is inadequate DNA repair. Frequent DNA repair is error-prone, particularly when the DNA repair machinery has been compromised by DNA damage. Chronic oxidative stress could saturate (overwhelm) DNA repair enzymes. New research indicates that CoQ10 not only can protect DNA from oxidative damage, but also enhances the activity of DNA repair enzymes. This animal study found that CoQ10 enrichment protects the DNA in lymphocytes from strand breaks, and in addition accelerates genetic repair. The researchers discovered that CoQ10 enhances the activity of DNA repair enzymes in supplemented animals.
Reversing bioenergetic decline
Linnane discovered a way to restore cellular vitality by improving energy production and resistance to stress in cells without intact mitochondrial DNA. In the early '90s he devised a technique to grow cells in culture after deleting their mitochondrial DNA. These cells were unable to assemble the machinery of cellular respiration. Instead, the growth process of these cells was supported by an alternative cellular energy source called glycolysis. Glycolysis produces energy from glucose, as happens most dramatically during strenuous muscular exercise. Normally, glycolysis relies upon the electron transport role of CoQ10 in the cellular respiratory chain. Linnane's insight was that CoQ10 could play the same role outside the mitochondria, in the outer cell membrane. He therefore added CoQ10 to the cell culture so that glycolysis, instead of cellular respiration, would sustain cell growth.
Linnane then extended his findings from cell cultures to lab animals. He tested his technique in rats treated with a drug that impairs mitochondrial function in a way that mimics aging. The drug halved the muscular force young rats were able to exert for an extended period, mirroring a decline in cellular bioenergetics. However, rats given CoQ10 or the CoQ10 analog CoQ10c along with the drug performed nearly as well as normal untreated rats. CoQ10 thus reversed the decline in cellular bioenergetics caused by mitochondrial impairment. Other studies by Linnane show how CoQ10 restores the capacity to respond to stress that is lost with age (see the sidebar “Restoring the Vital Response to Stress”).
“Dietary supplementation with coenzyme Q10 is thus indicated as a treatment to improve the quality of life of aged individuals and to provide protection against such age-related conditions as heart failure and neurodegenerative disease.”
From Anthony W. Linnane et al. (1998).
CoQ10 thus operates on three levels to stave off bioenergetic decline. First, CoQ10 helps protect mitochondrial DNA and the cellular respiratory chain from oxidative damage. Second, CoQ10 enhances cellular respiration, drawing maximal performance from the “bioenergy mosaic” that develops with age. Third, CoQ10 fuels an alternative energy source in the cell that helps compensate for declining cellular respiration.
The evolution of maximum lifespan in mammals
Each species of mammal has a known Maximum Lifespan Potential (MLSP). An intriguing line of research inspired by the free radical theory of aging suggests that the MLSP of each species corresponds to the level of a free radical called superoxide. Superoxide is a free radical formed from oxygen, especially when electrons leak out from the cellular respiratory chain. The lower the mitochondrial superoxide level in a given species, the longer that species lives. A similar relationship between superoxide and MLSP has also been found in fly species. While this does not necessarily mean that superoxide is a direct cause of aging, it does open up some fascinating lines of inquiry, albeit highly speculative ones.
In order to understand an insight into longevity that this research has provided, it is necessary to consider a fine point of animal physiology. In mammals, CoQ10 exists alongside the related form CoQ9. The proportions of CoQ10 and CoQ9 vary greatly between species. For example, rats and mice have mostly CoQ9, while rabbits, pigs and cows have mostly CoQ10 in heart cell mitochondria.
Antioxidant researchers Rajindar Sohal, Achim Lass and colleagues discovered that the higher the proportion of CoQ9 in a species, the more superoxide is generated in its heart mitochondria. The species with the highest proportions of CoQ10, on the other hand, have the lowest superoxide production in heart mitochondria and live the longest. As Lass and Sohal put it (1999), this finding is “consistent with the speculative notion that longevity co-evolved with a relative increase in the amounts of CoQ10.” In other words, the evolution of longer lifespan in mammals may be connected with the evolution of higher proportions of CoQ10.
There may be no meaningful way to test Sohal's hypothesis experimentally. He and his colleagues did make one attempt in which they altered the natural proportions of CoQ9 and CoQ10 in isolated submitochondrial particles from several species, then measured their rates of superoxide production. At normal physiological concentrations, superoxide levels remained the same; only at higher than normal concentrations did CoQ10 reduce superoxide generation. Thus the role of CoQ10 in evolution remains a thought-provoking though inconclusive hypothesis.
Programmed cell death
When cellular energy production declines in a mild gradual way, cells may adapt through compensatory systems such as glycolysis. However, when cellular energy levels drop more sharply, cells activate a process called “programmed cell death,” also called cellular suicide or apoptosis. Programmed cell death dismantles the cell in an orderly way with minimal damage to surrounding tissue. Linnane demonstrated this effect by culturing cells with a respiratory chain inhibitor that blocks cellular respiration. Within 12 to 18 hours, the cells underwent programmed cell death.
Linnane proposed that cellular suicide by energy-starved cells figures prominently in the pathology of age-associated disorders. Many lines of medical research point to widespread programmed cell death as a major factor in aging and degenerative diseases such as heart disease, cancer and neurodegeneration.
Recently published studies provide the first detailed picture of how programmed cell death works. It is now established that the mitochondria regulate this process, determining whether the cell lives or dies, and how it dies. While there are many pathways to cell death, the mitochondrial “decision to die” appears to spring largely from bioenergetic failure, oxidative stress and ion flows. New research demonstrates that CoQ10 directly inhibits the key event in programmed cell death, the opening of the mitochondrial “megachannel” that sets in motion the self-destruction of the cell (see “When Cellular Energy Declines” on Page 2).
Programmed cell death has been described for decades, but scientists are just beginning to unravel its molecular mechanisms. Programmed cell death is actuated by the opening of a channel in the inner membrane of the mitochondria called the "megachannel" (also called the permeability transition pore, or PTP). When the megachannel opens, the mitochondrial membrane becomes highly permeable and loses its electrical charge. Cell death-promoting factors from the mitochondrial inner membrane space are released into the cell. When this happens in a large enough proportion of the cell's mitochondria, the cell cannot survive. This process can lead either to programmed cell death, or to the more destructive cell death pathway called necrosis. What determines whether the megachannel opens and which path the dying cell takes?
We now know that programmed cell death is controlled by the mitochondria. It is thought that when a sudden bioenergetic catastrophe opens the megachannel before the cell can adapt, the cell undergoes violent necrotic death. On the other hand, when the megachannel opens gradually over a sufficient period of time, an orderly cellular suicide process unfolds instead.
A binding site for the CoQ10 family of compounds has been shown to regulate the opening of the mega-channel in rat liver and muscle cells. Moreover, groundbreaking new laboratory research shows that CoQ10 directly inhibits the opening of the megachannel.
Japanese research shows the visible effect of CoQ10 on cells under stress. Oxidative stress leads to programmed cell death, partly by damaging the cellular respiratory chain. As free radicals degrade the cell's metabolism regulatory mechanisms, DNA and proteins, the cell takes adaptive measures. The mitochondria typically enlarge or fuse to form "megamitochondria." Scientists speculate that this conserves energy or reduces free radical production. If oxidative stress subsides, the cell may return to normal. However, additional oxidative stress brings on programmed cell death.
Japanese scientists found that CoQ10 prevents these pathological changes. They gave one group of rats hydrazine, a drug that stimulates production of free radicals, for 7 to 8 days. They gave another group CoQ10 in addition to hydrazine. Hepatocytes (liver cells) from the hydrazine group showed "remarkably enlarged" mitochondria, while hepatocytes from the hydrazine plus CoQ10 group were only "slightly swollen," as illustrated below. The authors conclude that CoQ10 prevented megamitochondria formation by suppressing lipid peroxidation, and perhaps by preventing degradation of cellular respiration (uncoupling of oxygen consumption from ATP production).
Restoring the vital response to stress
The capacity to respond to stress declines with age. Younger patients, for example, recover faster from a heart attack or heart surgery than older patients. Linnane examined the cellular basis for recovery from cardiac stress, in collaboration with cardiology researcher Franklin Rosenfeldt and others. They compared heart tissue specimens from patients of different ages who had undergone heart surgery. They subjected the specimens to two stresses: hypoxia (oxygen deprivation) or simulated ischemia (interruption of blood flow). Following 30 minutes of stress, they let the specimens recover for another 30 minutes. The researchers then measured the contractile tension of the specimens to see how well they recovered from stress. As expected, tissues from older patients showed significantly less recovery following the stresses of hypoxia and ischemia. In agreement with Linnane's theory, they found a significant correlation between the integrity of mitochondrial DNA and the ability of tissues to recover from stress.
In a follow-up study, they incubated heart tissue in CoQ10 prior to simulated ischemia, then measured the contractile tension of the specimens. Tissue from patients seventy or more years of age recovered significantly less well from ischemia than tissue from younger patients, “but this difference was abolished by CoQ10” (Pepe S et al., 1999).
Next, Linnane and Rosenfeldt demonstrated that CoQ10 restores cardiovascular vitality in old rats. They measured two good indicators of cardiovascular vitality—how much work the heart can do during simulated aerobic exercise, and how well the heart recovers from exercise stress. Linnane and Rosenfeldt removed the intact hearts of young and aged rats and subjected the hearts to simulated exercise stress. They found that the hearts of aged rats are functionally debilitated when subjected to stress, showing markedly reduced efficiency and work capacity followed by poor recovery from stress as compared to young rats. Old rats treated with CoQ10 for six weeks regained full cardiovascular capacity: their hearts performed and recovered as well as those of young rats. In particular, hearts from CoQ10-treated old rats had four times the work capacity of untreated rats following exercise. CoQ10 treatment had no effect on the performance or recovery of hearts from young rats.
These studies comes as close as science can to a measurable definition of vitality. They show how the mitochondrial theory of aging explains stress response in heart tissue, and how CoQ10 restores energy and stress recovery in the aged heart to youthful levels. Mitochondrial aging depletes vitality, but mitochondrial rejuvenation may help to restore it.
A model of bioenergetic aging
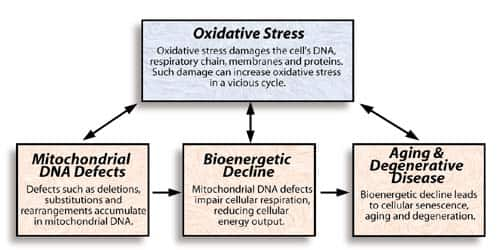 The bioenergetic theory of aging |
Figure 1. The series of boxes on the bottom shows how mitochondrial deterioration
can hasten aging and degeneration, as proposed by Linnane. Mitochondria are highly
susceptible to oxidative stress, which reinforces the other factors.
According to the free radical theory of aging, the buildup of oxidative stress and oxidative damage causes age-related degeneration. Since mitochondrial DNA and the cellular respiratory chain are highly susceptible to oxidative damage, this theory complements the bioenergetic theory of aging proposed by Linnane. Figure 2 illustrates how these theories might fit together.
When Cellular Energy Declines
Programmed cell death is a well-orchestrated process of cellular self-destruction. As the cell shrinks and then fragments, its organelles remain relatively intact and enclosed by membranes. Neighboring cells or macrophages safely digest the fragments. By contrast, in necrotic cell death the cell swells and ruptures, organelles disintegrate, and inflammation tends to occur.
References
Adachi K et al. A deletion of mitochondrial DNA in murine doxorubicin-induced cardiotoxicity. 1993. Biochem Biophys Res Comm 195: 945-951.
Adachi K et al. Suppression of the hydrazine-induced formation of megamitochondria in the rat liver by coenzyme Q10. 1995. Toxicol Pathol 23: 667-676.
Alleva R et al. Supplementation with coenzyme Q10 protects DNA against oxidative damage and enhances DNA repair enzyme activity. 2000. Free Radic Biol Med 29, Suppl 1: S80.
Ames BN et al. Mitochondrial decay in aging. 1995. Biochim Biophys Acta 1271: 165-170.
Arbustini E et al. Mitochondrial DNA mutations and mitochondrial abnormalities in dilated cardiomyopathy. 1998. Am J Pathol 153: 1501-1510.
Cavalli LR et al. Mutagenesis, tumorigenicity, and apoptosis: are the mitochondria involved? 1998. Mutat Res 398: 19-26.
“Cellular Nutrition for Vitality and Longevity,” LIFE EXTENSION magazine, April 2000, pp. 24-28.
DiMauro S et al. Mitochondria in neuromuscular disorders. 1998. Biochim Biophys Acta 1366: 199-210.
Esposito LA et al. Mitochondrial disease in mouse results in increased oxidative stress. 1999. Proc Natl Acad Sci USA 96: 4820-4825.
Fontaine E et al. A ubiquinone-binding site regulates the mitochondrial permeability transition pore. 1998. J Biol Chem 273: 25734-25740.
Fontaine E et al. Regulation of the permeability transition pore in skeletal muscle mitochondria. 1998. J Biol Chem 273: 12662-12668.
Geromel V et al. The consequences of a mild respiratory chain deficiency on substrate competitive oxidation in human mitochondria. 1997. Biochem Biophys Res Comm 236: 643-646.
Karbowski M et al. Free radical-induced megamitochondria formation and apoptosis. 1999. Free Radic Biol Med 26: 396-409.
Kopsidas G et al. An age-associated correlation between cellular bioenergy decline and mtDNA rearrangements in human skeletal muscle. 1998. Mutat Res 421: 27-36.
Kovalenko SA et al. Tissue-specific distribution of multiple mitochondrial DNA rearrangements during human aging. 1998. Ann NY Acad Sci 854: 171-181.
Ku HH et al. Relationship between mitochondrial superoxide and hydrogen peroxide production and longevity of mammalian species. 1993. Free Radic Biol Med 15: 621-627.
Lass A et al. Mitochondrial ubiquinone homologues, superoxide radical generation, and longevity in different mammalian species. 1997. J Biol Chem 272: 19199-19204.
Lass A et al. Comparisons of coenzyme Q bound to mitochondrial membrane proteins among different mammalian species. 1999. Free Radic Biol Med 27: 220-226.
Linnane AW et al. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. 1989. Lancet 1: 642-645.
Linnane AW et al. The universality of bioenergetic disease and amelioration with redox therapy. 1995. Biochim Biophys Acta 1271: 191-194.
Linnane AW et al. The universality of bioenergetic disease. Age-associated cellular bioenergetic degradation and amelioration therapy. 1998. Ann NY Acad Sci 854: 202-213.
Martinucci S et al. Ca2+-reversible inhibition of the mitochondrial megachannel by ubiquinone analogues. 2000. FEBS Lett 480: 89-94.
Michikawa Y et al. Aging-dependent large accumulation of point mutations in the human mtDNA control region for replication. 1999. Science. 286: 774-9.
Ozawa T. Genetic and functional changes in mitochondria associated with aging. 1997. Physiol Rev 77: 425-464.
Pepe S et al. Coenzyme Q10 normalizes impaired post-ischemic contractile recovery of aged human myocardium in vitro. 1998. Circulation 98, Suppl: 3602.
Richter C et al. Control of apoptosis by the cellular ATP level. 1996. FEBS Lett 378: 107-110.
Rosenfeldt FL et al. Response of the human myocardium to hypoxia and ischemia declines with age. 1998. Ann NY Acad Sci 854: 489-490.
Rowland MA et al. Coenzyme Q10 treatment improves the tolerance of the senescent myocardium to pacing stress in the rat. 1998. Cardiovasc Res 40: 165-173.
Sohal RS et al. Mitochondrial superoxide and hydrogen peroxide generation, protein oxidative damage, and longevity in different species of flies. 1995. Free Radic Biol Med 19: 499-504.
Susin SA et al. Mitochondria as regulators of apoptosis: doubt no more. 1998. Biochim Biophys Acta 1366: 151-165.
Turker MS. Somatic cell mutations: can they provide a link between aging and cancer? 2000. Mech Ageing Dev 117: 1-19.
Wallace DC. Mitochondrial diseases in man and mouse. 1999. Science 283: 1482-1488.
Wallace DC et al. Mitochondrial DNA mutations in human degenerative diseases and aging. 1995. Biochim Biophys Acta 1271: 141-151.
Walter L et al. Three classes of ubiquinone analogs regulate the mitochondrial permeability transition pore through a common site. 2000. J Biol Chem 275: 29521-29527.
Wei YH. Oxidative stress and mitochondrial DNA mutations in human aging. 1998. Proc Soc Exp Biol Med 217: 53-63.
Wei YH et al. Simultaneous increase of mitochondrial DNA deletions and lipid peroxidation in human aging. 1996. Proc NY Acad Sci 786: 24-43.
Zhang C et al. Varied prevalence of age-associated mitochondrial DNA deletions in different species and tissues: a comparison between human and rat. 1997. Biochem Biophys Res Comm 230: 630-635.
Wolvetang EJ et al. Mitochondrial respiratory chain inhibitors induce apoptosis. 1994. FEBS Lett 339: 40-44.
Wellness
Specialists
1-800-226-2370 - This service is FREE
7:30 AM - 12 AM (ET) Mon-Fri | 9 AM - 12 AM (ET) Sat-Sun