Life Extension Magazine®
Two months ago, the New England Journal of Medicine published two studies showing that moderately lowering homocysteine blood levels did not reduce heart attack risk in those with existing cardiac or vascular disease. The following statement was made in an editorial that accompanied these two articles: “. . . the results lead to the unequivocal conclusion that there is no clinical benefit to the use of folic acid and vitamin B12 (with or without vitamin B6) in patients with established vascular disease.”1 The media then boldly proclaimed: “Hope Abandoned for Benefit to Lowering Homocysteine” Contrary to these negative opinions, the two studies in the New England Journal of Medicine confirm what Life Extension long ago published about vascular disease and what steps are required to achieve optimal homocysteine control. As a reader of Life Extension magazine, you have a front row seat to a raging debate that could very well affect how many Americans will develop heart disease, stroke, osteoporosis, Alzheimer’s disease, blindness, depression, and other disorders associated with excess homocysteine. Homocysteine was first theorized to cause vascular disease when autopsy results of young people revealed atherosclerotic plaque in those with very high homocysteine levels. Over a 37-year period, doctors uncovered startling evidence linking elevated homocysteine to increased risks of heart attack,2-36 stroke,37-50 and other disorders in aging adults. In addition to human epidemiological data, scientists have identified specific mechanisms by which homocysteine causes the most common age-related diseases. One of the New England Journal of Medicine authors summed up these toxic mechanisms by stating: “ . . . homocysteine is an atherogenic determinate that promotes oxidant stress, inflammation, thrombosis, endothelial dysfunction, and cell proliferation.”1 Based on the description above, excess homocysteine would appear to be linked to virtually every human degenerative disease. If this is the case, then why did the same author state that reducing homocysteine had no effect in preventing heart attack?
Homocysteine May Not Have Been Reduced EnoughScientific studies dating back to the early 1990s indicate that optimal homocysteine levels should not exceed 9-10 micromoles per liter (µmol/L),51 and ideally should be even lower than that, perhaps under 7 µmol/L for optimal risk reduction.52 One epidemiological study demonstrated that homocysteine levels above 10 µmol/L are associated with an increase in heart attack risk.22 Another study showed that homocysteine levels as low as 9 carry long-term danger, with cardiac risk escalating more sharply when homocysteine levels are at 15 or greater.17 Still another (Japanese) study showed that those with a homocysteine level below 7 were much less likely to suffer a stroke than patients with homocysteine levels higher than 11.51 The New England Journal of Medicine recently published two studies showing no benefit to lowering homocysteine in those with pre-existing vascular disease. In the first study, baseline homocysteine levels of 12.2 µmol/L were reduced to a mean of 9.7 over a two-year period. Study participants in the active group were given one daily supplement that consisted of 2.5 mg of folic acid, 1 mg of vitamin B12, and 50 mg of vitamin B6.53 In the second New England Journal of Medicine study, the baseline homocysteine level of 13 µmol/L was reduced to a mean of 9.6 after three years. One active group in this study received only 40 mg of vitamin B6, while other groups received 0.8 mg of folic acid and 0.4 mg of B12 with and without 40 mg of B6.54 Life Extension long ago advocated that members take aggressive steps to keep homocysteine levels below 7-8 µmol/L.55-57 The rationale was based on an extrapolation of the existing published studies relating to homocysteine blood levels and heart attack risk.
For instance, an earlier study17 in the New England Journal of Medicine looked at mortality in coronary artery disease patients and found the following: As the table above shows, homocysteine blood levels between 9 and 15 µmol/L doubled mortality rates compared to values less than 9, while homocysteine above 15 increased mortality by an astounding 6.47 times compared to levels below 9! The two recent studies published in the New England Journal of Medicine failed to reduce homocysteine to the levels that the Life Extension Foundation long ago stated were needed to reduce heart attack risk. In fact, these two recent studies failed to reduce homocysteine to the less than 9 µmol/L level that had previously been shown in the same medical journal to confer benefit. The question still begs, however, as to why moderate homocysteine reduction did not result in at least some reduction in heart attack risk. The answers become obvious as the study’s design methods are uncovered. Unhealthy Study SubjectsTo participate in the first study that used modest doses of nutrients to moderately reduce homocysteine levels, one had to have “a history of vascular disease (coronary, cerebrovascular, or peripheral vascular) or diabetes and additional risk factors for atherosclerosis.” To qualify for the second study that used lower potencies of nutrients, participants had to first suffer “an acute myocardial infarction,” more commonly known as a heart attack, within seven days of enrollment in the study. Based on the enrollment criteria, the study subjects had to have significant pre-existing vascular impairment in order to participate. In this article, you will read why the use of these unhealthy subjects virtually guaranteed that this study would fail. Next, however, we need to emphasize that. . .
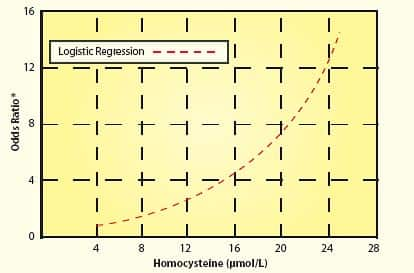
The Study Period Was Too Short!Based on the enrollment criteria, the study subjects had already suffered significant arterial damage over most of their lifetime. The study design expectation was that within two or three years, taking modest doses of only three nutrients would somehow protect the test subjects against future vascular events. Please note the word “within” when describing the study period. While the first study lasted two years and the second study lasted a little over three years, any participant who encountered a vascular event even one day after the study’s commencement was considered a statistic. In other words, if a study participant suffered any kind of vascular disease after they took even one dose of B vitamins, then that person was listed as a “failure,” meaning that he or she filled a statistical column showing no benefit to B vitamins as far as this study was concerned. Remember that these were high-risk participants with pre-existing vascular disease. It is absurd to expect that moderately lowering homocysteine within a very short time period would result in a miraculous disease reduction in these unhealthy individuals. As any enlightened person knows, atherosclerosis does not develop overnight. Initial arterial lesions are sometimes seen in teenagers. The process of inner arterial wall degradation leading to atherosclerosis lasts for many decades. When one has pre-existing vascular disease—as was the case for most of these study subjects—the arteries are severely damaged and the risk of future arterial-related diseases is high. For people with pre-existing arterial disease, protecting against a future heart attack or stroke requires extraordinary effort. Swallowing one modest-dose daily supplement after you have developed significant vascular disease is not going to do it! Baseline Homocysteine Levels May Not Have Been High EnoughWe discussed before that the two studies failed to adequately reduce homocysteine levels to optimal safe ranges. While that is one significant flaw, another defect in these two studies was that the subjects’ homocysteine levels were not particularly high to begin with. To clarify this point, while some studies show that homocysteine levels above 7-9 µmol/L increase vascular disease risk, the most lethal effect occurs when levels exceed 13-15. The subjects chosen for these two studies had baseline homocysteine levels of 12.2 and 13 on average, which were reduced to 9.7 and 9.6, respectively. Existing data indicate that there may not have been much of a statistical difference between the study subjects’ baseline levels and their homo-cysteine levels two and three years later. The following chart using data from a study published by the American Heart Association helps explain why the baseline homocysteine levels in these two studies may not have been high enough.58 As one can see, cardiac risk increases incrementally with rising homocysteine. Generally speaking, when the odds ratio exceeds 2.0 or 2.5 in epidemiology studies, scientists become concerned that risk increases. In this chart, a homocysteine level of about 13 µmo/L corresponds to an odds ratio of about 2.0 to 2.5. From the chart, the risk associated with a homocysteine level of 13-15 is 4 to 5 times greater compared to the risk associated with a homocysteine level of 7-8.It should be noted that this chart uses logistic regression to describe heart attack risk in relationship to increasing homocysteine levels.
| ||||||||||||||||||||||||||||

Other Cardiac Risk Factors IgnoredThe design of these studies took into account conventional risk factors for vascular disease such as cholesterol, low-density lipoprotein (LDL), high-density lipoprotein (HDL), triglycerides, glucose, body mass index, and blood pressure. For example, the average fasting glucose level at baseline was 128.8 mg/dL in one group receiving the B vitamins and 125.7 mg/dL in the placebo group. Both groups clearly had poor glucose control (pre-diabetic, at a minimum). The independent vascular disease risk factors that were ignored include C-reactive protein, fibrinogen, fasting insulin, hemoglobin A1C, and free testosterone. Imbalances of any one of these “other” risk factors would have almost certainly negated the benefit that would be expected from a modest reduction in homocysteine. The evidence linking C-reactive protein to vascular disease risk is overwhelming.65-82 In one scientific study after another, elevated levels of C-reactive protein are shown to be strong independent markers for determining who is likely to suffer a heart attack or stroke. Without factoring in proven risk factors such as C-reactive protein (along with fibrinogen,83-93 free testosterone,94-97 and others), it is difficult to know whether more subjects in the B-vitamin group required additional therapies beyond modest homocysteine reduction. Other than the most basic conventional medical interventions, study subjects continued following the unhealthy lifestyles that may have induced their vascular disease, including cigarette smoking and poor eating habits. It is quite a leap of faith for the designers of these studies to have expected that modest homocysteine reduction would overcome dangerous lifestyles that are proven to cause cardiovascular diseases.
This Study Has Nothing to Do with Healthy PeopleDespite the media proclaiming that the homocysteine theory of vascular disease has been discredited, the fact is that the authors of the studies themselves state that the findings pertain only to those with existing vascular disease. Nowhere in the studies do they suggest that healthy people seeking to prevent atherosclerosis may not benefit from reducing their homocysteine levels. The authors of all of the studies went to great length in explaining why homocysteine is such a dangerous amino acid.
In today’s world of sound bites, the message from a study of people with serious disease somehow is distorted into a news story that is supposed to pertain to those who do not even have the disease. For instance, we know that selenium does not cure cancer, but it certainly appears that it prevents certain cancers.98 The fact that selenium is not an effective cancer treatment does not mean that those seeking to prevent cancer should not take it. These two studies showed that modest homocysteine reduction by itself did not prevent cardiac events in those with pre-existing disease. This has no relevance for those seeking to maintain healthy arterial function by keeping their homocysteine in the lower ranges before they develop severe arterial disease. In fact, one of these same groups of researchers is initiating a five-year study of healthy individuals to ascertain whether modest homocysteine reduction lowers vascular disease risk. The headline-hungry media ignored the fact that homocysteine-lowering therapies are still being researched when proclaiming the homocysteine theory of arterial disease to be “dead.”
Lethal Misconceptions About AtherosclerosisThe hottest buzzword in cardiovascular research today is endothelial dysfunction, a term that describes the structural and functional damage to the inner arterial wall that so often results in athero-sclerosis. Atherosclerosis is the cause of most heart attacks and strokes, yet many doctors still do not understand that this artery-blocking process is both initiated and accelerated via the destructive endothelial dysfunction process. The inner lining of the arterial wall is made up of a thin layer of cells called the endothelium. The endothelium protects the smooth muscle in the middle wall of the artery from direct contact with the blood. The endothelial barrier is important to arterial health because many blood components are highly toxic to the artery’s smooth muscle (that lies directly beneath the endothelium). When these damaging blood components attack the artery’s smooth muscle, the process of atherosclerosis is initiated. Among the blood components that are damaging to the inner lining of arteries are glucose,99,100 homocysteine,101-122 low-density lipoprotein (LDL),123-131 free radicals,109,132-136 and pro-inflammatory cytokines.4,137-147 To protect the artery’s elastic smooth muscle against these damaging agents, it is critical to maintain an intact and properly functioning endothelium. The endothelium can be disrupted at an early age because of poor health habits such as cigarette smoking, poor diet, and nutrient deficiencies. The aging process itself results in endothelial dysfunction that can be caused by a number of known risk factors. The inner wall of diseased arteries is a battleground where multiple pathological reactions take place. Endothelial dysfunction occurring in damaged arteries provides fertile soil for the seeds of atherosclerosis. To expect a modest reduction of homocysteine to reverse this devastating process—especially after cardiac disease has already manifested—flies in the face of all that has been learned about the underlying causes of heart attack, stroke, and other vascular diseases. What Causes Endothelial Dysfunction?High blood pressure,148-153 elevated LDL,123-31 low HDL,154-156 cigarette smoking,157-163 diabetes,164-169 obesity,170-172 and lack of exercise173-175 all contribute to endothelial dysfunction and the subsequent development of atherosclerosis. Additional endothelial-damaging factors include excess glucose,99,100 insulin,176 iron,177-179 homocysteine,103-122 fibrinogen,85-93 and C-reactive protein,65-82 as well as low free testosterone (in men).94-97 Homocysteine is dangerous because it can induce initial injury to the endothelium, then facilitate the oxidation of the fat/LDL that accumulates beneath the damaged endothelium, and finally contribute to the abnormal accumulation of blood components around the atherosclerotic lesion. Fibrinogen is a clotting factor that accumulates at the site of the endothelial lesion, contributing to plaque buildup or participating in the blockage of an artery by a blood clot after an unstable atherosclerotic plaque ruptures. Glucose at even high-normal levels may accelerate the glycation process that causes arterial stiffening, while high-normal fasting insulin inflicts direct damage to the endothelium. High levels of iron promote LDL oxidation in the damaged endothelium, while low levels of testosterone appear to interfere with normal endothelial function. C-reactive protein not only is an inflammatory marker, but also directly damages the endothelium. Chronic inflammation, as evidenced by persistent high levels of C-reactive protein, causes initial injuries to the endothelium and also accelerates the progression of existing atherosclerotic lesions. Clearly, then, the degenerative process of endothelial dysfunction has multiple underlying causes. A modest reduction of homocysteine alone is not going to overcome all of the other risk factors involved in arterial degeneration. Nor is modest homocysteine reduction going to reverse a lifetime of cumulative damage to the arterial wall. Yet that is what these two recent studies were designed to demonstrate.
Why These Two Studies Were Doomed to FailHomocysteine is one of many causes of endothelial dysfunction. Endothelial dysfunction is known to occur in otherwise healthy people with elevations in fasting homocysteine ranging from 15 to 35 µmol/L.180 Furthermore, in otherwise healthy people, impaired endothelial function is seen with small increases in plasma homocysteine (2-3 µmol/L), even when blood homocysteine does not rise above the upper limit of “normal” (15 µmol/L).181 This means that even relatively low circulating levels of homocysteine can damage the arterial wall! Despite the documented evidence of homocysteine’s adverse effects on normal endothelial function, expecting a significant drop in cardiovascular risk in a group of patients with severe pre-existing endothelial dysfunction with a modest dose of B vitamins is quite a stretch. Yet, that was exactly the assumption made in the two recent studies.
For example, in one of the homocysteine-lowering trials, patients had to have suffered a heart attack within seven days of enrolling in the study. These patients would have already had significant endothelial impairment, and it is well known that the risk of sudden death is greatly increased during the time after a heart attack. For example, a study published in the New England Journal of Medicine evaluated 14,609 patients who suffered heart attacks. The risk of dying was highest in the first 30 days after a heart attack. In fact, 83% of all patients who died suddenly did so in the first 30 days after hospital discharge, and risk persisted for up to two years.182 Clearly, these patients had a high degree of endothelial dysfunction to begin with, and were a very sick group of patients. Assuming that B vitamins alone would have a major impact in this very sick patient population—with severe, pre-existing endothelial dysfunction and at high risk of sudden death—is a major design flaw. When one steps back and looks at what was done in these studies, it should be no surprise that cardiac risks were not lowered in the B-vitamin groups. Based on everything we know about arterial disease, it would have been impossible to reverse the severe arterial damage in these patients merely by reducing homocysteine levels a few points within the normal mid-range. The study subjects’ baseline homocysteine levels were not severely high. This means it is likely that their arterial disease was caused by some of the other known risk factors. For instance, compared to the placebo arm, a statistically significantly higher number of people in the B-vitamin group of one study were treated with warfarin (Coumadin®). Could this point to some unknown factor at baseline that increased cardiac risk in the B-vitamin group? A Windfall for Drug CompaniesPharmaceutical companies profit more from sales of cardiac drugs than from any other class of medication. With all the media hype about homocysteine not being a cardiac risk factor, many doctors will advise their patients to discontinue their B-vitamin supplements. This is particularly unfortunate for the coronary artery disease patients with homocysteine readings of 15 µmol/L and above. While these two studies do not relate to cardiac patients with these higher homocysteine readings, hurried doctors (who neglected to read the actual studies) may uniformly advise all their cardiac patients to discontinue B-vitamin supplements. Uninformed consumers will reduce their B-vitamin consumption based on media reports that homocysteine is no longer considered a cardiac risk factor. All of this adds up to a financial windfall for drug companies, which stand to sell their expensive cardiac drugs to millions of new heart attack patients who become victims of homocysteine-induced atherosclerosis. Excess homocysteine causes more than just heart attacks. Osteoporosis,183-185 depression,186-189 and Alzheimer’s disease,190-200 as well as sharply increased risks of stroke,37-50 are linked with elevated homocysteine. If even a small percentage of the American public discontinues its B-vitamin supplements, the demand for prescription antidepressants, anti-Alzheimer’s drugs (acetylcholinesterase inhibitors), and bone-building prescription medications such as the bisphosphonates is likely to skyrocket. Discrediting low-cost nutrients that have been shown to reduce disease risk is great news for companies that sell drugs to those who fall ill. In a forthcoming issue, we will follow up with a feature article by Dr. William Davis discussing novel nutritional strategies that you can use to lower high homocysteine levels and gain truly effective control of this potentially lethal compound. | ||||||||||
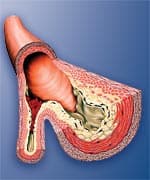
Protect Yourself Against the Epidemic of Vascular DiseaseThe number-one killer in the United States is atherosclerosis, which causes most heart attacks and strokes.59 Doctors remain confused as to how this artery-blocking process occurs and overlook the numerous independent factors that can inflict severe damage to the arterial wall. Many of the underlying causes of atherosclerosis are readily detectable with annual blood tests. Once the risk factors that predispose one to a heart attack or stroke are identified, corrective actions can be initiated. What follows are the 10 most important blood tests one should have at least once a year to evaluate cardiac health:
Some members are able to get blood tests from their own doctors. One problem is that even when doctors order all the blood tests requested, the phlebotomist may fail to check off the appropriate codes on the laboratory requisition form, or may not properly draw the blood. When the results come back incomplete, another blood draw becomes necessary, thus inconveniencing the patient. Even today, many doctors still refuse to prescribe important cardiovascular risk blood tests such as fibrinogen and C-reactive protein. Life Extension resolved this problem 10 years ago by offering blood tests directly to its members. Once a year, we reduce our everyday low prices. Until May 31, 2006, blood test prices are discounted so that members can obtain complete blood evaluations at a fraction of the price charged by commercial laboratories. The Male Panel and Female Panel provide the most important blood tests that can identify risk factors before overt disease manifests. At a commercial laboratory, the price of the tests that make up the Male Panel is $1,164. During our annual blood test super sale, members can obtain these same tests for only $224 . . . a savings of over 80%! Similar savings are available on the Female Life Extension Panel. The normal member price for the fibrinogen and hemoglobin A1C tests is $62. Until May 31, 2006, we are discounting this price for both of these tests to $25 when the Male Panel or Female Panel is ordered. This means that members can obtain a more complete evaluation of their cardiac risk factors at a lower cost than ever before. For example, a Male Life Extension Panel or Female Life Extension Panel plus fibrinogen plus hemoglobin A1C costs only $249 during the blood test super sale. To order your own blood tests at these sale prices, call 1-800-208-3444 or order online. |
| References |
| 1. Loscalzo J. Homocysteine trials—clear outcomes for complex reasons. N Engl J Med. 2006 Mar 14. 2. Available at: www.lifeextension.com/magazine/mag2003/refs/nov. Accessed April 4, 2006. 3. Kazemi MB, Eshraghian K, Omrani GR, Lankarani KB, Hosseini E. Homocysteine level and coronary artery disease. Angiology. 2006 Jan;57(1):9-14. 4. Rasouli ML, Nasir K, Blumenthal RS, et al. Plasma homocysteine predicts progression of atherosclerosis. Atherosclerosis. 2005 Jul;181(1):159-65. 5. Toole JF, Malinow MR, Chambless LE, et al. Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction, and death: the Vitamin Intervention for Stroke Prevention (VISP) randomized controlled trial. JAMA. 2004 Feb 4;291(5):565-75. 6. Retterstol L, Paus B, Bohn M, et al. Plasma total homocysteine levels and prognosis in patients with previous premature myocardial infarction: a 10-year follow-up study. J Intern Med. 2003 Mar;253(3):284-92. 7. Adachi H, Hirai Y, Fujiura Y, et al. Plasma homocysteine levels and atherosclerosis in Japan: epidemiological study by use of carotid ultrasonography. Stroke. 2002 Sep;33(9):2177-81. 8. Kuan YM, Dear AE, Grigg MJ. Homocysteine: an aetiological contributor to peripheral vascular arterial disease. ANZ J Surg. 2002 Sep;72(9):668-71. 9. Auer J, Berent R, Weber T, Lassnig E, Eber B. Homocysteine and cardiovascular risk. Wien Med Wochenschr. 2001;151(1-2):25-8. 10. Beaudeux JL, Jacob N, Giral P, Foglietti MJ, Bruckert E. New non-lipidic biological markers of atherosclerosis. Ann Med Interne (Paris). 2001 Apr;152(3):169-79. 11. Aronow WS. Association between plasma homocysteine and vascular atherosclerotic disease in older persons. Prev Cardiol. 2000;3(2):89-91. 12. Dzielinska Z, Kadziela J, Sitkiewicz D, et al. Elevated levels of homocysteine in plasma as a risk factor for coronary artery disease. Pol Arch Med Wewn. 2000 Jul;104(1):345-53. 13. Giles WH, Croft JB, Greenlund KJ, Ford ES, Kittner SJ. Association between total homocyst(e)ine and the likelihood for a history of acute myocardial infarction by race and ethnicity: results from the Third National Health and Nutrition Examination Survey. Am Heart J. 2000 Mar;139(3):446-53. 14. Eikelboom JW, Lonn E, Genest J, Jr., Hankey G, Yusuf S. Homocyst(e)ine and cardiovascular disease: a critical review of the epidemiologic evidence. Ann Intern Med. 1999 Sep 7;131(5):363-75. 15. Magott M. Homocysteine as a nonlipid factor in the pathogenesis of atherosclerosis. Postepy Hig Med Dosw. 1998;52(3):259-67. 16. Refsum H, Ueland PM, Nygard O, Vollset SE. Homocysteine and cardiovascular disease. Annu Rev Med. 1998;49:31-62. 17. Nygard O, Nordrehaug JE, Refsum H, et al. Plasma homocysteine levels and mortality in patients with coronary artery disease. N Engl J Med. 1997 Jul 24;337(4):230-6. 18. Fallest-Strobl PC, Koch DD, Stein JH, McBride PE. Homocysteine: a new risk factor for atherosclerosis. Am Fam Physician. 1997 Oct 15;56(6):1607-6. 19. Nehler MR, Taylor LM, Jr., Porter JM. Homocysteinemia as a risk factor for atherosclerosis: a review. Cardiovasc Surg. 1997 Dec;5(6):559-67. 20. Mayer EL, Jacobsen DW, Robinson K. Homocysteine and coronary atherosclerosis. J Am Coll Cardiol. 1996 Mar 1;27(3):517-27. 21. Malinow MR, Nieto FJ, Szklo M, Chambless LE, Bond G. Carotid artery intimal-medial wall thickening and plasma homocyst(e)ine in asymptomatic adults. The Atherosclerosis Risk in Communities Study. Circulation. 1993 Apr;87(4):1107-13. 22. Stampfer MJ, Malinow MR, Willett WC, et al. A prospective study of plasma homocyst(e)ine and risk of myocardial infarction in US physicians. JAMA. 1992 Aug 19;268(7):877-81. 23. Taylor LM, Jr., DeFrang RD, Harris EJ, Jr., Porter JM. The association of elevated plasma homocyst(e)ine with progression of symptomatic peripheral arterial disease. J Vasc Surg. 1991 Jan;13(1):128-36. 24. Clarke R, Daly L, Robinson K, et al. Hyperhomocysteinemia: an independent risk factor for vascular disease. N Engl J Med. 1991 Apr 25;324(17):1149-55. 25. Ubbink JB, Vermaak WJ, Bennett JM, et al. The prevalence of homocysteinemia and hypercholesterolemia in angiographically defined coronary heart disease. Klin Wochenschr. 1991 Aug 16;69(12):527-34. 26. Genest JJ, Jr., McNamara JR, Salem DN, et al. Plasma homocyst(e)ine levels in men with premature coronary artery disease. J Am Coll Cardiol. 1990 Nov;16(5):1114-9. 27. Malinow MR, Kang SS, Taylor LM, et al. Prevalence of hyperhomocyst(e)inemia in patients with peripheral arterial occlusive disease. Circulation. 1989 Jun;79(6):1180-8. 28. Israelsson B, Brattstrom LE, Hultberg BL. Homocysteine and myocardial infarction. Atherosclerosis. 1988 Jun;71(2-3):227-33. 29. Olszewski AJ, Szostak WB. Homocysteine content of plasma proteins in ischemic heart disease. Atherosclerosis. 1988 Feb;69(2-3):109-13. 30. Kang SS, Wong PW, Cook HY, Norusis M, Messer JV. Protein-bound homocyst(e)ine. A possible risk factor for coronary artery disease. J Clin Invest. 1986 May;77(5):1482-6. 31. Boers GH, Smals AG, Trijbels FJ, et al. Heterozygosity for homocystinuria in premature peripheral and cerebral occlusive arterial disease. N Engl J Med. 1985 Sep 19;313(12):709-15. 32. Murphy-Chutorian DR, Wexman MP, Grieco AJ, et al. Methionine intolerance: a possible risk factor for coronary artery disease. J Am Coll Cardiol. 1985 Oct;6(4):725-30. 33. Wilcken DE, Wilcken B. The pathogenesis of coronary artery disease. A possible role for methionine metabolism. J Clin Invest. 1976 Apr;57(4):1079-82. 34. McCully KS, Wilson RB. Homocysteine theory of arteriosclerosis. Atherosclerosis. 1975 Sep;22(2):215-27. 35. McCully KS. Homocystine, atherosclerosis and thrombosis: implications for oral contraceptive users. Am J Clin Nutr. 1975 May;28(5):542-9. 36. McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol. 1969 Jul;56(1):111-28. 37. Casas JP, Bautista LE, Smeeth L, Sharma P, Hingorani AD. Homocysteine and stroke: evidence on a causal link from mendelian randomisation. Lancet. 2005 Jan 15;365(9455):224-32. 38. Sachdev PS, Valenzuela MJ, Brodaty H, et al. Homocysteine as a risk factor for cognitive impairment in stroke patients. Dement Geriatr Cogn Disord. 2003;15(3):155-62. 39. Tanne D, Haim M, Goldbourt U, et al. Prospective study of serum homocysteine and risk of ischemic stroke among patients with preexisting coronary heart disease. Stroke. 2003 Mar;34(3):632-6. 40. Kelly PJ, Rosand J, Kistler JP, et al. Homocysteine, MTHFR 677C—>T polymorphism, and risk of ischemic stroke: results of a meta-analysis. Neurology. 2002 Aug 27;59(4):529-36. 41. bdel-Raheem MM, Hebert B, Potti A, Koka VK, Danielson BD. Hyperhomocysteinemia and the risk of thromboembolic phenomenon in patients with chronic renal failure. Thromb Res. 2002 Feb 15;105(4):299-302. 42. Matsui T, Arai H, Yuzuriha T, et al. Elevated plasma homocysteine levels and risk of silent brain infarction in elderly people. Stroke. 2001 May;32(5):1116-9. 43. Available at: www.medscape.com/viewarticle/408377_print. Accessed April 4, 2006. 44. Bostom AG, Rosenberg IH, Silbershatz H, et al. Nonfasting plasma total homocysteine levels and stroke incidence in elderly persons: the Framingham Study. Ann Intern Med. 1999 Sep 7;131(5):352-5. 45. Perry IJ, Refsum H, Morris RW, et al. Prospective study of serum total homocysteine concentration and risk of stroke in middle-aged British men. Lancet. 1995 Nov 25;346(8987):1395-8. 46. Brattstrom L, Lindgren A, Israelsson B, et al. Hyperhomocysteinaemia in stroke: prevalence, cause, and relationships to type of stroke and stroke risk factors. Eur J Clin Invest. 1992 Mar;22(3):214-21. 47. Coull BM, Malinow MR, Beamer N, et al. Elevated plasma homocyst(e)ine concentration as a possible independent risk factor for stroke. Stroke. 1990 Apr;21(4):572-6. 48. Brattstrom LE, Hardebo JE, Hultberg BL. Moderate homocysteinemia—a possible risk factor for arteriosclerotic cerebrovascular disease. Stroke. 1984 Nov-Dec;15(6):1012-6. 49. Graeber JE, Slott JH, Ulane RE, Schulman JD, Stuart MJ. Effect of homocysteine and homocystine on platelet and vascular arachidonic acid metabolism. Pediatr Res. 1982 Jun;16(6):490-3. 50. Ganz P, Vita JA. Testing endothelial vasomotor function: nitric oxide, a multipotent molecule. Circulation. 2003 Oct 28;108(17):2049-53. 51. Iso H, Moriyama Y, Sato S, et al. Serum total homocysteine concentrations and risk of stroke and its subtypes in Japanese. Circulation. 2004 Jun 8;109(22):2766-72. 52. Spence JD. Patients with atherosclerotic vascular disease: how low should plasma homocyst(e)ine levels go? Am J Cardiovasc Drugs. 2001;1(2):85-9. 53. Lonn E, Held C, Arnold JM, et al. Rationale, design and baseline characteristics of a large, simple, randomized trial of combined folic acid and vitamins B6 and B12 in high-risk patients: the Heart Outcomes Prevention Evaluation (HOPE)-2 trial. Can J Cardiol. 2006 Jan;22(1):47-53. 54. Bonaa KH, Njolstad I, Ueland PM, et al. Homocysteine Lowering and Cardiovascular Events after Acute Myocardial Infarction. N Engl J Med. 2006 Mar 12. 55. Available at: www.lifeextension.com/magazine/mag2003/nov2003_awsi_01.html. Accessed April 4, 2006. 56. Available at: http://www.lifeextension.com/magazine/mag2004/may2004_report_blood_01.htm. Accessed April 4, 2006. 57. Available at: http://www.lifeextension.com/magazine/mag99/mar99-report2.html. Accessed April 4, 2006. 58. Available at: http://circ.ahajournals.org/ content/vol92/issue10/images/large/hc2250986001.jpeg. Accessed April 4, 2006. 59. Available at: http://www.cdc.gov/nchs/fastats/lcod.htm. Accessed April 4, 2006. 60. Available at: http://www.ninds.nih.gov/disorders/stroke/knowstroke.htm. Accessed April 4, 2006. 61. Available at: http://www.ninds.nih.gov/disorders/stroke/knowstroke.htm. Accessed April 4, 2006. 62. Available at: http://www.lifeextension.com/magazine/mag2003/nov2003_awsi_01.htm. Accessed April 4, 2006. 63. Available at: http://www.lifeextension.com/magazine/mag2004/may2004_report_blood_01.htm. Accessed April 4, 2006. 64. Available at: http://www.lifeextension.com/magazine/mag99/mar99-report2.html. Accessed April 4, 2006. 65. Auer J, Berent R, Lassnig E, Eber B. C-reactive protein and coronary artery disease. Jpn Heart J. 2002 Nov;43(6):607-19. 66. Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 2002 Nov 14;347(20):1557-65. 67. Wang TJ, Larson MG, Levy D, et al. C-reactive protein is associated with subclinical epicardial coronary calcification in men and women: the Framingham Heart Study. Circulation. 2002 Sep 3;106(10):1189-91. 68. Bermudez EA, Ridker PM. C-reactive protein, statins, and the primary prevention of atherosclerotic cardiovascular disease. Prev Cardiol. 2002;5(1):42-6. 69. Virmani R, Burke AP, Kolodgie FD, Farb A. Vulnerable plaque: the pathology of unstable coronary lesions. J Interv Cardiol. 2002 Dec;15(6):439-46. 70. Rifai N, Ridker PM. Inflammatory markers and coronary heart disease. Curr Opin Lipidol. 2002 Aug;13(4):383-9. 71. Zairis MN, Papadaki OA, Manousakis SJ, et al. C-reactive protein and multiple complex coronary artery plaques in patients with primary unstable angina. Atherosclerosis. 2002 Oct;164(2):355-9. 72. Ridker PM. High-sensitivity C-reactive protein: potential adjunct for global risk assessment in the primary prevention of cardiovascular disease. Circulation. 2001 Apr 3;103(13):1813-8. 73. Di NM, Papa F, Bocola V. Prognostic influence of increased C-reactive protein and fibrinogen levels in ischemic stroke. Stroke. 2001 Jan;32(1):133-8. 74. Ridker PM, Stampfer MJ, Rifai N. Novel risk factors for systemic atherosclerosis: a comparison of C-reactive protein, fibrinogen, homocysteine, lipoprotein(a), and standard cholesterol screening as predictors of peripheral arterial disease. JAMA. 2001 May 16;285(19):2481-5. 75. Rifai N. C-reactive protein and coronary heart disease: diagnostic and therapeutic implications for primary prevention. Cardiovasc Toxicol. 2001;1(2):153-7. 76. Higuchi M, Castelli JB, Aiello VD, et al. Great amount of C.pneumoniae in ruptured plaque vessel segments at autopsy. A comparative study with stable plaques. Arq Bras Cardiol. 2000 Feb;74(2):149-51. 77. Pasceri V, Willerson JT, Yeh ET. Direct proinflammatory effect of C-reactive protein on human endothelial cells. Circulation. 2000 Oct 31;102(18):2165-8. 78. Mendall MA, Strachan DP, Butland BK, et al. C-reactive protein: relation to total mortality, cardiovascular mortality and cardiovascular risk factors in men. Eur Heart J. 2000 Oct;21(19):1584-90. 79. Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000 Mar 23;342(12):836-43. 80. Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Plasma concentration of C-reactive protein and risk of developing peripheral vascular disease. Circulation. 1998 Feb 10;97(5):425-8. 81. Ridker PM, Buring JE, Shih J, Matias M, Hennekens CH. Prospective study of C-reactive protein and the risk of future cardiovascular events among apparently healthy women. Circulation. 1998 Aug 25;98(8):731-3. 82. Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997 Apr 3;336(14):973-9. 83. Drouet L, Bal dit SC. Is fibrinogen a predictor or a marker of the risk of cardiovascular events? Therapie. 2005 Mar;60(2):125-36. 84. Coppola G, Rizzo M, Abrignani MG, et al. Fibrinogen as a predictor of mortality after acute myocardial infarction: a forty-two-month follow-up study. Ital Heart J. 2005 Apr;6(4):315-22. 85. Danesh J, Lewington S, Thompson SG, et al. Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: an individual participant meta-analysis. JAMA. 2005 Oct 12;294(14):1799-809. 86. Acevedo M, Foody JM, Pearce GL, Sprecher DL. Fibrinogen: associations with cardiovascular events in an outpatient clinic. Am Heart J. 2002 Feb;143(2):277-82. 87. Bots ML, Elwood PC, Salonen JT, et al. Level of fibrinogen and risk of fatal and non-fatal stroke. EUROSTROKE: a collaborative study among research centres in Europe. J Epidemiol Community Health. 2002 Feb;56 Suppl 1i14-8. 88. de Maat MP. Effects of diet, drugs, and genes on plasma fibrinogen levels. Ann NY Acad Sci. 2001;936:509-21. 89. Lindahl B, Toss H, Siegbahn A, Venge P, Wallentin L. Markers of myocardial damage and inflammation in relation to long-term mortality in unstable coronary artery disease. FRISC Study Group. Fragmin during Instability in Coronary Artery Disease. N Engl J Med. 2000 Oct 19;343(16):1139-47. 90. Maresca G, Di BA, Marchioli R, Di MG. Measuring plasma fibrinogen to predict stroke and myocardial infarction: an update. Arterioscler Thromb Vasc Biol. 1999 Jun;19(6):1368-77. 91. Ma J, Hennekens CH, Ridker PM, Stampfer MJ. A prospective study of fibrinogen and risk of myocardial infarction in the Physicians’ Health Study. J Am Coll Cardiol. 1999 Apr;33(5):1347-52. 92. Behar S. Lowering fibrinogen levels: clinical update. BIP Study Group. Bezafibrate Infarction Prevention. Blood Coagul Fibrinolysis. 1999 Feb;10 Suppl 1S41-3. 93. Thompson SG, Kienast J, Pyke SD, Haverkate F, van de Loo JC. Hemostatic factors and the risk of myocardial infarction or sudden death in patients with angina pectoris. European Concerted Action on Thrombosis and Disabilities Angina Pectoris Study Group. N Engl J Med. 1995 Mar 9;332(10):635-41. 94. Jones RD, Nettleship JE, Kapoor D, Jones HT, Channer KS. Testosterone and atherosclerosis in aging men: purported association and clinical implications. Am J Cardiovasc Drugs. 2005;5(3):141-54. 95. Channer KS, Jones TH. Cardiovascular effects of testosterone: implications of the “male menopause”? Heart. 2003 Feb;89(2):121-2. 96. Malkin CJ, Pugh PJ, Jones RD, Jones TH, Channer KS. Testosterone as a protective factor against atherosclerosis—immunomodulation and influence upon plaque development and stability. J Endocrinol. 2003 Sep;178(3):373-80. 97. English KM, Mandour O, Steeds RP, et al. Men with coronary artery disease have lower levels of androgens than men with normal coronary angiograms. Eur Heart J. 2000 Jun;21(11):890-4. 98. Clark LC, Combs GF, Jr., Turnbull BW, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA. 1996 Dec 25;276(24):1957-63. 99. Stochmal E, Szurkowska M, Czarnecka D, et al. Association of coronary atherosclerosis with insulin resistance in patients with impaired glucose tolerance. Acta Cardiol. 2005 Jun;60(3):325-31. 100. Ceriello A. Impaired glucose tolerance and cardiovascular disease: the possible role of post-prandial hyperglycemia. Am Heart J. 2004 May;147(5):803-7. 101. Zeng XK, Guan YF, Remick DG, Wang X. Signal pathways underlying homocysteine-induced production of MCP-1 and IL-8 in cultured human whole blood. Acta Pharmacol Sin. 2005 Jan;26(1):85-91. 102. Zeng XK, Remick DG, Wang X. Homocysteine induces production of monocyte chemoattractant protein-1 and interleukin-8 in cultured human whole blood. Acta Pharmacol Sin. 2004 Nov;25(11):1419-25. 103. Hassan A, Hunt BJ, O’Sullivan M, et al. Homocysteine is a risk factor for cerebral small vessel disease, acting via endothelial dysfunction. Brain. 2004 Jan;127(Pt 1):212-9. 104. Devlin AM, Arning E, Bottiglieri T, et al. Effect of Mthfr genotype on diet-induced hyperhomocysteinemia and vascular function in mice. Blood. 2004 Apr 1;103(7):2624-9. 105. Ungvari Z, Csiszar A, Edwards JG, et al. Increased superoxide production in coronary arteries in hyperhomocysteinemia: role of tumor necrosis factor-alpha, NAD(P)H oxidase, and inducible nitric oxide synthase. Arterioscler Thromb Vasc Biol. 2003 Mar 1;23(3):418-24. 106. Loscalzo J. Oxidant stress: a key determinant of atherothrombosis. Biochem Soc Trans. 2003 Oct;31(Pt 5):1059-61. 107. Faraci FM. Hyperhomocysteinemia: a million ways to lose control. Arterioscler Thromb Vasc Biol. 2003 Mar 1;23(3):371-3. 108. Symons JD, Mullick AE, Ensunsa JL, Ma AA, Rutledge JC. Hyperhomocysteinemia evoked by folate depletion: effects on coronary and carotid arterial function. Arterioscler Thromb Vasc Biol. 2002 May 1;22(5):772-80. 109. Eberhardt RT, Forgione MA, Cap A, et al. Endothelial dysfunction in a murine model of mild hyperhomocyst(e)inemia. J Clin Invest. 2000 Aug;106(4):483-91. 110. Folsom AR, Nieto FJ, McGovern PG, et al. Prospective study of coronary heart disease incidence in relation to fasting total homocysteine, related genetic polymorphisms, and B vitamins: the Atherosclerosis Risk in Communities (ARIC) study. Circulation. 1998 Jul 21;98(3):204-10. 111. Woo KS, Chook P, Lolin YI, et al. Hyperhomocyst(e)inemia is a risk factor for arterial endothelial dysfunction in humans. Circulation. 1997 Oct 21;96(8):2542-4. 112. Bots ML, Launer LJ, Lindemans J, Hofman A, Grobbee DE. Homocysteine, atherosclerosis and prevalent cardiovascular disease in the elderly: The Rotterdam Study. J Intern Med. 1997 Oct;242(4):339-47. 113. Montalescot G, Ankri A, Chadefaux-Vekemans B, et al. Plasma homocysteine and the extent of atherosclerosis in patients with coronary artery disease. Int J Cardiol. 1997 Aug 8;60(3):295-300. 114. Lentz SR, Sobey CG, Piegors DJ, et al. Vascular dysfunction in monkeys with diet-induced hyperhomocyst(e)inemia. J Clin Invest. 1996 Jul 1;98(1):24-9. 115. Verhoef P, Stampfer MJ, Buring JE, et al. Homocysteine metabolism and risk of myocardial infarction: relation with vitamins B6, B12, and folate. Am J Epidemiol. 1996 May 1;143(9):845-59. 116. Robinson K, Mayer EL, Miller DP, et al. Hyperhomocysteinemia and low pyridoxal phosphate. Common and independent reversible risk factors for coronary artery disease. Circulation. 1995 Nov 15;92(10):2825-30. 117. Arnesen E, Refsum H, Bonaa KH, et al. Serum total homocysteine and coronary heart disease. Int J Epidemiol. 1995 Aug;24(4):704-9. 118. Berwanger CS, Jeremy JY, Stansby G. Homocysteine and vascular disease. Br J Surg. 1995 Jun;82(6):726-31. 119. Tsai JC, Perrella MA, Yoshizumi M, et al. Promotion of vascular smooth muscle cell growth by homocysteine: a link to atherosclerosis. Proc Natl Acad Sci USA. 1994 Jul 5;91(14):6369-73. 120. Fryer RH, Wilson BD, Gubler DB, Fitzgerald LA, Rodgers GM. Homocysteine, a risk factor for premature vascular disease and thrombosis, induces tissue factor activity in endothelial cells. Arterioscler Thromb. 1993 Sep;13(9):1327-33. 121. Harker LA, Harlan JM, Ross R. Effect of sulfinpyrazone on homocysteine-induced endothelial injury and arteriosclerosis in baboons. Circ Res. 1983 Dec;53(6):731-9. 122. Wall RT, Rubenstein MD, Cooper SL. Studies on the cellular basis of atherosclerosis: the effects of atherosclerosis risk factors on platelets and the vascular endothelium. Diabetes. 1981;30(Suppl 2):39-43. 123. Maggi FM, Raselli S, Grigore L, et al. Lipoprotein remnants and endothelial dysfunction in the postprandial phase. J Clin Endocrinol Metab. 2004 Jun;89(6):2946-50. 124. Shatrov VA, Brune B. Induced expression of manganese superoxide dismutase by non-toxic concentrations of oxidized low-density lipoprotein (oxLDL) protects against oxLDL-mediated cytotoxicity. Biochem J. 2003 Sep 1;374(Pt 2):505-11. 125. Sainani GS, Sainani R. Homocysteine and its role in the pathogenesis of atherosclerotic vascular disease. J Assoc Physicians India. 2002 May;50 Suppl16-23. 126. Albert CM, Ma J, Rifai N, Stampfer MJ, Ridker PM. Prospective study of C-reactive protein, homocysteine, and plasma lipid levels as predictors of sudden cardiac death. Circulation. 2002 Jun 4;105(22):2595-9. 127. Laaksonen R, Janatuinen T, Vesalainen R, et al. High oxidized LDL and elevated plasma homocysteine contribute to the early reduction of myocardial flow reserve in healthy adults. Eur J Clin Invest. 2002 Nov;32(11):795-802. 128. Dardik R, Varon D, Tamarin I, et al. Homocysteine and oxidized low density lipoprotein enhanced platelet adhesion to endothelial cells under flow conditions: distinct mechanisms of thrombogenic modulation. Thromb Haemost. 2000 Feb;83(2):338-44. 129. Voutilainen S, Morrow JD, Roberts LJ, et al. Enhanced in vivo lipid peroxidation at elevated plasma total homocysteine levels. Arterioscler Thromb Vasc Biol. 1999 May;19(5):1263-6. 130. De CR, Lenzi S. The role of LDL in the origin and progression of atherosclerosis: pathobiological concepts on the origin and development of atherosclerotic lesions and the role of the endothelium. G Ital Cardiol. 1998 Feb;28(2):158-67. 131. Drexel H, Amann FW, Beran J, et al. Plasma triglycerides and three lipoprotein cholesterol fractions are independent predictors of the extent of coronary atherosclerosis. Circulation. 1994 Nov;90(5):2230-5. 132. Johansen JS, Harris AK, Rychly DJ, Ergul A. Oxidative stress and the use of antioxidants in diabetes: linking basic science to clinical practice. Cardiovasc Diabetol. 2005 Apr 29;4(1):5. 133. Schulz E, Anter E, Keaney JF, Jr. Oxidative stress, antioxidants, and endothelial function. Curr Med Chem. 2004 May;11(9):1093-104. 134. Maxwell SR. Coronary artery disease—free radical damage, antioxidant protection and the role of homocysteine. Basic Res Cardiol. 2000;95 Suppl 1I65-71. 135. Kanani PM, Sinkey CA, Browning RL, et al. Role of oxidant stress in endothelial dysfunction produced by experimental hyperhomocyst(e)inemia in humans. Circulation. 1999 Sep 14;100(11):1161-8. 136. Loscalzo J. The oxidant stress of hyperhomocyst(e)inemia. J Clin Invest. 1996 Jul 1;98(1):5-7. 137. Li JJ, Chen JL. Inflammation may be a bridge connecting hypertension and atherosclerosis. Med Hypotheses. 2005;64(5):925-9. 138. Nurk E, Tell GS, Vollset SE, et al. Changes in lifestyle and plasma total homocysteine: the Hordaland Homocysteine Study. Am J Clin Nutr. 2004 May;79(5):812-9. 139. Tentolouris C, Tousoulis D, Antoniades C, et al. Endothelial function and proinflammatory cytokines in patients with ischemic heart disease and dilated cardiomyopathy. Int J Cardiol. 2004 Apr;94(2-3):301-5. 140. Fan J, Watanabe T. Inflammatory reactions in the pathogenesis of atherosclerosis. J Atheroscler Thromb. 2003;10(2):63-71. 141. Pradhan AD, Manson JE, Rossouw JE, et al. Inflammatory biomarkers, hormone replacement therapy, and incident coronary heart disease: prospective analysis from the Women’s Health Initiative observational study. JAMA. 2002 Aug 28;288(8):980-7. 142. Blake GJ, Ridker PM. Inflammatory mechanisms in atherosclerosis: from laboratory evidence to clinical application. Ital Heart J. 2001 Nov;2(11):796-800. 143. Jialal I, Devaraj S. Inflammation and atherosclerosis: the value of the high-sensitivity C-reactive protein assay as a risk marker. Am J Clin Pathol. 2001 Dec;116 SupplS108-15. 144. Lowe GD. The relationship between infection, inflammation, and cardiovascular disease: an overview. Ann Periodontol. 2001 Dec;6(1):1-8. 145. Aleman G, Tovar AR, Torres N. Homocysteine metabolism and risk of cardiovascular diseases: importance of the nutritional status on folic acid, vitamins B6 and B12. Rev Invest Clin. 2001 Mar;53(2):141-51. 146. Van der Meide PH, Schellekens H. Cytokines and the immune response. Biotherapy. 1996;8(3-4):243-9. 147. Dudman NP, Guo XW, Gordon RB, Dawson PA, Wilcken DE. Human homocysteine catabolism: three major pathways and their relevance to development of arterial occlusive disease. J Nutr. 1996 Apr;126(4 Suppl):1295S-300S. 148. Chang HJ, Chung J, Choi SY, et al. Endothelial dysfunction in patients with exaggerated blood pressure response during treadmill test. Clin Cardiol. 2004 Jul;27(7):421-5. 149. Higashi Y, Yoshizumi M. Exercise and endothelial function: role of endothelium-derived nitric oxide and oxidative stress in healthy subjects and hypertensive patients. Pharmacol Ther. 2004 Apr;102(1):87-96. 150. Rodriguez-Porcel M, Lerman LO, Herrmann J et al. Hypercholesterolemia and hypertension have synergistic deleterious effects on coronary endothelial function. Arterioscler Thromb Vasc Biol. 2003 May 1;23(5):885-91. 151. Tu L, Wei W, Liu X, Deng Y, Yu S. Endothelial function and carotid artery wall thickening in patients with early essential hypertension. J Tongji Med Univ. 1999;19(4):288-90, 303. 152. Johnson LK, Longenecker JP, Fajardo LF. Differential radiation response of cultured endothelial cells and smooth myocytes. Anal Quant Cytol. 1982 Sep;4(3):188-98. 153. Sutton-Tyrrell K, Bostom A, Selhub J, Zeigler-Johnson C. High homocysteine levels are independently related to isolated systolic hypertension in older adults. Circulation. 1997 Sep 16;96(6):1745-9. 154. Calabresi L, Gomaraschi M, Franceschini G. Endothelial protection by high-density lipoproteins: from bench to bedside. Arterioscler Thromb Vasc Biol. 2003 Oct 1;23(10):1724-31. 155. Spieker LE, Sudano I, Hurlimann D, et al. High-density lipoprotein restores endothelial function in hypercholesterolemic men. Circulation. 2002 Mar 26;105(12):1399-402. 156. Toikka JO, Ahotupa M, Viikari JS, et al. Constantly low HDL-cholesterol concentration relates to endothelial dysfunction and increased in vivo LDL-oxidation in healthy young men. Atherosclerosis. 1999 Nov 1;147(1):133-8. 157. Ikonomidis I, Lekakis J, Vamvakou G, Andreotti F, Nihoyannopoulos P. Cigarette smoking is associated with increased circulating proinflammatory and procoagulant markers in patients with chronic coronary artery disease: effects of aspirin treatment. Am Heart J. 2005 May;149(5):832-9. 158. Esen AM, Barutcu I, Acar M, et al. Effect of smoking on endothelial function and wall thickness of brachial artery. Circ J. 2004 Dec;68(12):1123-6. 159. Wanner A, Campos MA, Mendes E. Airway blood flow reactivity in smokers. Pulm Pharmacol Ther. 2006 Jan 13; [Epub ahead of print] 160. Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol. 2004 May 19;43(10):1731-7. 161. Poreba R, Skoczynska A, Derkacz A. Effect of tobacco smoking on endothelial function in patients with coronary arteriosclerosis. Pol Arch Med Wewn. 2004 Jan;111(1):27-36. 162. Puranik R, Celermajer DS. Smoking and endothelial function. Prog Cardiovasc Dis. 2003 May;45(6):443-58. 163. Chrysohoou C, Panagiotakos DB, Pitsavos C, et al. The associations between smoking, physical activity, dietary habits and plasma homocysteine levels in cardiovascular disease-free people: the ‘ATTICA’ study. Vasc Med. 2004 May;9(2):117-23. 164. O’Callaghan P, Meleady R, Fitzgerald T, Graham I. Smoking and plasma homocysteine. Eur Heart J. 2002 Oct;23(20):1580-6. 165. Targher G, Bertolini L, Zoppini G, Zenari L, Falezza G. Increased plasma markers of inflammation and endothelial dysfunction and their association with microvascular complications in Type 1 diabetic patients without clinically manifest macroangiopathy. Diabet Med. 2005 Aug;22(8):999-1004. 166. Jarvisalo MJ, Raitakari M, Toikka JO, et al. Endothelial dysfunction and increased arterial intima-media thickness in children with type 1 diabetes. Circulation. 2004 Apr 13;109(14):1750-5. 167. Vlassara H, Cai W, Crandall J, et al. Inflammatory mediators are induced by dietary glycotoxins, a major risk factor for diabetic angiopathy. Proc Natl Acad Sci USA. 2002 Nov 26;99(24):15596-601. 168. Najemnik C, Sinzinger H, Kritz H. Endothelial dysfunction, atherosclerosis and diabetes. Acta Med Austriaca. 1999;26(5):148-53. 169. Hoogeveen EK, Kostense PJ, Beks PJ, et al. Hyperhomocysteinemia is associated with an increased risk of cardiovascular disease, especially in non-insulin-dependent diabetes mellitus: a population-based study. Arterioscler Thromb Vasc Biol. 1998 Jan;18(1):133-8. 170. Bakker SJ, IJzerman RG, Teerlink T, et al. Cytosolic triglycerides and oxidative stress in central obesity: the missing link between excessive atherosclerosis, endothelial dysfunction, and beta-cell failure? Atherosclerosis. 2000 Jan;148(1):17-21. 171. Yu YR, Li HL, Yu HL, Wang C, Pu S. The relationship between insulin resistance and endothelium-dependent vasodilatation in obese subjects. Zhonghua Yi Xue Za Zhi. 2003 Sep 10;83(17):1467-70. 172. Blann AD, Bushell D, Davies A, et al. von Willebrand factor, the endothelium and obesity. Int J Obes Relat Metab Disord. 1993 Dec;17(12):723-5. 173. Edwards DG, Schofield RS, Lennon SL, et al. Effect of exercise training on endothelial function in men with coronary artery disease. Am J Cardiol. 2004 Mar 1;93(5):617-20. 174. Mitu F, Mitu M. Physical exercise and vascular endothelium. Rev Med Chir Soc Med Nat Iasi. 2003 Jul;107(3):487-93. 175. Gokce N, Vita JA, Bader DS, et al. Effect of exercise on upper and lower extremity endothelial function in patients with coronary artery disease. Am J Cardiol. 2002 Jul 15;90(2):124-7. 176. Muis MJ, Bots ML, Bilo HJ, et al. High cumulative insulin exposure: a risk factor of atherosclerosis in type 1 diabetes? Atherosclerosis. 2005 Jul;181(1):185-92. 177. Howes PS, Zacharski LR, Sullivan J, Chow B. Role of stored iron in atherosclerosis. J Vasc Nurs. 2000 Dec;18(4):109-14. 178. de VB, Marx JJ. Iron, atherosclerosis, and ischemic heart disease. Arch Intern Med. 1999 Jul 26;159(14):1542-8. 179. Chau LY. Iron and atherosclerosis. Proc Natl Sci Counc Repub China B. 2000 Oct;24(4):151-5. 180. Bellamy MF, McDowell IF, Ramsey MW, et al. Oral folate enhances endothelial function in hyperhomocysteinaemic subjects. Eur J Clin Invest. 1999 Aug;29(8):659-62. 181. Chambers JC, Obeid OA, Kooner JS. Physiological increments in plasma homocysteine induce vascular endothelial dysfunction in normal human subjects. Arterioscler Thromb Vasc Biol. 1999 Dec;19(12):2922-7. 182. Solomon SD, Zelenkofske S, McMurray JJ, et al. Sudden death in patients with myocardial infarction and left ventricular dysfunction, heart failure, or both. N Engl J Med. 2005 Jun 23;352(25):2581-8. 183. van Meurs JB, Dhonukshe-Rutten RA, Pluijm SM, et al. Homocysteine levels and the risk of osteoporotic fracture. N Engl J Med. 2004 May 13;350(20):2033-41. 184. McLean RR, Jacques PF, Selhub J, et al. Homocysteine as a predictive factor for hip fracture in older persons. N Engl J Med. 2004 May 13;350(20):2042-9. 185. Morris MS, Jacques PF, Selhub J. Relation between homocysteine and B-vitamin status indicators and bone mineral density in older Americans. Bone. 2005 Aug;37(2):234-42. 186. Bjelland I, Tell GS, Vollset SE, Refsum H, Ueland PM. Folate, vitamin B12, homocysteine, and the MTHFR 677C->T polymorphism in anxiety and depression: the Hordaland Homocysteine Study. Arch Gen Psychiatry. 2003 Jun;60(6):618-26. 187. Naismith S, Hickie I, Ward PB, et al. Caudate nucleus volumes and genetic determinants of homocysteine metabolism in the prediction of psychomotor speed in older persons with depression. Am J Psychiatry. 2002 Dec;159(12):2096-8. 188. Fava M, Borus JS, Alpert JE, et al. Folate, vitamin B12, and homocysteine in major depressive disorder. Am J Psychiatry. 1997 Mar;154(3):426-8. 189. Bottiglieri T, Laundy M, Crellin R, et al. Homocysteine, folate, methylation, and monoamine metabolism in depression. J Neurol Neurosurg Psychiatry. 2000 Aug;69(2):228-32. 190. Religa D, Styczynska M, Peplonska B, et al. Homocysteine, apolipoproteine E and methylenetetrahydrofolate reductase in Alzheimer’s disease and mild cognitive impairment. Dement Geriatr Cogn Disord. 2003;16(2):64-70. 191. Morris MS. Homocysteine and Alzheimer’s disease. Lancet Neurol. 2003 Jul;2(7):425-8. 192. Selley ML. Increased concentrations of homocysteine and asymmetric dimethylarginine and decreased concentrations of nitric oxide in the plasma of patients with Alzheimer’s disease. Neurobiol Aging. 2003 Nov;24(7):903-7. 193. Ho PI, Ortiz D, Rogers E, Shea TB. Multiple aspects of homocysteine neurotoxicity: glutamate excitotoxicity, kinase hyperactivation and DNA damage. J Neurosci Res. 2002 Dec 1;70(5):694-702. 194. Seshadri S, Beiser A, Selhub J, et al. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N Engl J Med. 2002 Feb 14;346(7):476-83. 195. McIlroy SP, Dynan KB, Lawson JT, Patterson CC, Passmore AP. Moderately elevated plasma homocysteine, methylenetetrahydrofolate reductase genotype, and risk for stroke, vascular dementia, and Alzheimer disease in Northern Ireland. Stroke. 2002 Oct;33(10):2351-6. 196. Joosten E. Homocysteine, vascular dementia and Alzheimer’s disease. Clin Chem Lab Med. 2001 Aug;39(8):717-20. 197. Miller JW. Homocysteine, Alzheimer’s disease, and cognitive function. Nutrition. 2000 Jul;16(7-8):675-7. 198. McCaddon A, Davies G, Hudson P, Tandy S, Cattell H. Total serum homocysteine in senile dementia of Alzheimer type. Int J Geriatr Psychiatry. 1998 Apr;13(4):235-9. 199. Gottfries CG, Lehmann W, Regland B. Early diagnosis of cognitive impairment in the elderly with the focus on Alzheimer’s disease. J Neural Transm. 1998;105(8-9):773-86. 200. Snowdon DA, Greiner LH, Mortimer JA, et al. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA. 1997 Mar 12;277(10):813-7. |
Wellness
Specialists
1-800-226-2370 - This service is FREE
7:30 AM - 12 AM (ET) Mon-Fri | 9 AM - 12 AM (ET) Sat-Sun