
Blood Clot Prevention
Blood Clot Prevention
Last Section Update: 06/2024
Contributor(s): Maureen Williams, ND; Shayna Sandhaus, PhD
Table of Contents
- Overview
- Introduction
- Hemostasis
- Thrombosis
- Dietary and Lifestyle Considerations
- Nutrients
- Causes, Risk Factors & Associated Conditions
- Blood Clot Signs & Symptoms
- Diagnosis of Blood Clot-Related Conditions
- Treatment of Blood Clots & Thrombosis
- Monitoring Blood Clot Therapy
- Novel & Emerging Strategies to Prevent & Treat Thrombosis
- Update History
- References
1 Overview
Summary and Quick Facts for Blood Clot Prevention
- If you are over 50, the greatest threat to your health is the formation of abnormal blood clots in your arteries and veins.
- This protocol discusses blood clot symptoms, clotting mechanisms, and how the body keeps them under control, as well as how they become dysregulated, increasing the risk of blood clots.
- The medical approach to blood clot prevention in high-risk individuals relies on antiplatelet and anticoagulant drugs, sometimes called blood thinners, which can cause serious side effects due to bleeding. In severe cases requiring hospitalization, IV administration of emergency thrombolytics (“clot busters”) may be used.
- Eating a more plant-based, unprocessed diet and getting regular exercise can help lower the risk of blood clots.
- Certain nutrients, such as omega-3 fatty acids, ginkgo, and garlic, may help prevent blood clots.
What are Blood Clots?
Normal blood clots are an essential part of the healing process, both inside blood vessels and at external injury sites. However, if a blood clot blocks blood flow to organs or tissues, it can be very dangerous. Abnormal blood clots (thrombosis) are the most common cause of heart attacks and strokes.
High-risk individuals are generally treated with “blood thinner” drugs including antiplatelet drugs like aspirin and clopidogrel or anticoagulants like heparin, warfarin, and dabigatran. While these drugs can reduce the risk of developing a clot, they fail to address many underlying risk factors and can have serious side effects.
Nutrients like fish oil, ginkgo, garlic, and cocoa flavanols may help prevent thrombosis and related complications.
What Dietary and Lifestyle Changes Can Help Prevent Blood Clots?
- Eat a plant-based unprocessed diet rich in fruits, vegetables, and unsaturated fats, such as a Mediterranean style or DASH diet
- Exercise regularly
- Manage stress
What Nutrients May Help Prevent Blood Clots?
- Fish oil. Fish oil and its omega-3 fatty acids lower the risk of thrombotic events such as heart attack and stroke, as well as deep vein thrombosis.
- Ginkgo. Ginkgo may improve platelet and blood vessel function, and has been shown in preclinical studies to inhibit platelet activation and aggregation and promote the breakdown of blood clots.
- Garlic. Garlic has been shown to promote cardiovascular health in many human studies. One of the observed benefits is garlic’s ability to reduce platelet aggregation, which may help protect against excessive blood clotting.
- Cocoa flavanols. Cocoa intake is correlated with cardiovascular health and lower risk of conditions that raise thrombosis risk. Clinical trials show cocoa flavanols can reduce platelet aggregation.
- Coenzyme Q10. Clinical trials have shown coenzyme Q10 supplementation can lower levels of markers of thrombotic risk, improve blood vessel function, and reduce cardiovascular risk.
- Pycnogenol. Pycnogenol, extracted from French maritime pine bark, has been shown to reduce fluid build-up and blood clots in people with a history of venous thrombosis.
- Probiotics. Certain probiotic bacteria, especially the Lactobacillus reuteri NCIMB 30242 strain, have been found to improve markers of thrombosis risk.
- Nattokinase. Nattokinase has demonstrated the ability to increase the breakdown of blood clots. Together with pycnogenol, nattokinase was found to reduce leg edema and venous blood clots in long-haul air travelers.
- B vitamins. Vitamin B12 and folic acid deficiencies cause high homocysteine levels, which have been associated with increased atherosclerosis, stroke, arterial clots, and venous thrombosis risk. Vitamin B3 (niacin) may also reduce thrombosis risk by inhibiting platelet aggregation and supporting blood clot breakdown.
- Other natural interventions that may help prevent blood clots and improve cardiovascular health include green tea extract, pomegranate, saffron, quercetin, ginger, and guavirova.
What are Some Risk Factors and Conditions Associated with Blood Clots?
- Older age
- History of a previous thrombotic event
- Smoking
- Chronic venous disease
- Atherosclerosis
- Abnormal lipid levels
- Hypertension
- Type 2 diabetes
- Obesity
- Sleep apnea
- Prolonged inactivity
- Hospitalization or surgery
- Infection, transplant, and transfusion reactions
- Pregnancy
- Cancer
- Use of high-dose oral estrogens (eg, hormone replacement therapy or a high-dose birth control pill)
How are Blood Clots Treated?
- Emergency thrombolytic (“clot buster”) agents and sonothrombolysis (clot-breaking ultrasound)
- Some cases may be amenable to emergency surgery to remove a clot (thrombectomy)
- Blood thinners, including antiplatelet drugs (eg, aspirin and clopidogrel), mainly for arterial thrombosis, and anticoagulants (eg, heparin, warfarin, and dabigatran)
What are Some Emerging Therapies for Blood Clots?
- New methods of sonothrombolysis
- Canakinumab
- Repurposed drugs: statins, colchicine, and metformin
2 Introduction
Blood clotting is an important part of hemostasis, the balance of anti-coagulation and pro-coagulation mechanisms that preserves normal blood flow throughout the body. In healthy conditions, blood clots, which includes venous and arterial clots, form to prevent blood loss after an injury to a blood vessel. However, in some circumstances, hemostatic signaling becomes dysregulated and excessive blood clotting or abnormal clots that obstruct blood flow can form.1,2 Depending on where these clots occur, they can cause dangerous or even life-threatening complications.3
Blood clots can form in veins (venous clots), interfering with the return of blood from tissues to the heart, or arteries (arterial clots), interfering with movement of oxygenated blood from the heart to tissues and organs throughout the body. A blood clot attached to a vessel wall is known as a thrombus and one that breaks free and travels in the blood is called an embolus. Pulmonary embolism, venous thromboembolism (VTE), and post-thrombotic syndrome are dangerous complications of venous blood clots, while serious complications of arterial blood clots include heart attack and stroke. Complications of blood clots are the most common cause of death in developed countries and are responsible for one of every four deaths worldwide.3,4
Smoking, older age, obesity and metabolic syndrome, family history, a sedentary lifestyle, infection, and certain health conditions and medications increase the risk of developing an abnormal blood clot.3,5 Injury to a vessel, such as due to surgery or trauma, can also trigger thrombosis while immobility during recovery can also be a provoking factor. In addition, the risk of venous thrombosis or VTE is higher in women during pregnancy and in those using oral birth control pills or oral estrogen-containing postmenopausal hormone replacement therapy.3
Blood clots are diagnosed using imaging such as specialized ultrasound, venography (an x-ray taken after a dye is injected into the vein), magnetic resonance imaging (MRI), or computed tomography (CT) techniques.6,7 Blood tests may be helpful in determining if a clot exists and for monitoring the blood’s propensity to form clots.2 Once a blood clot is found, it may require immediate treatment with thrombolytic medication or clot busters, mechanical destruction using ultrasound, or surgical removal.8,9 Individuals with a high risk of arterial clots may be treated with one or more long-term antiplatelet blood thinner medications like aspirin and clopidogrel (Plavix). Those at high risk of recurrent venous thrombosis or VTE may receive long-term anticoagulant therapy using warfarin or a direct oral anticoagulant (DOAC). While effective for reducing blood clots, these medications can all cause serious bleeding side effects.3 Importantly, they also do nothing to address the underlying conditions that contribute to thrombosis risk.
Because of the potentially devastating consequences of blood clots, it is critical to reduce their risk using dietary, lifestyle, and nutrient strategies. In addition to addressing modifiable risk factors, a plant-based, minimally processed diet, regular exercise, and stress management are the cornerstones of blood clot prevention and should be considered primary interventions. In addition, a number of nutrients, including ginseng, ginkgo, garlic, and cocoa have been found in clinical trials to reduce the likelihood of thrombosis.
3 Hemostasis
Hemostasis and Blood Clots
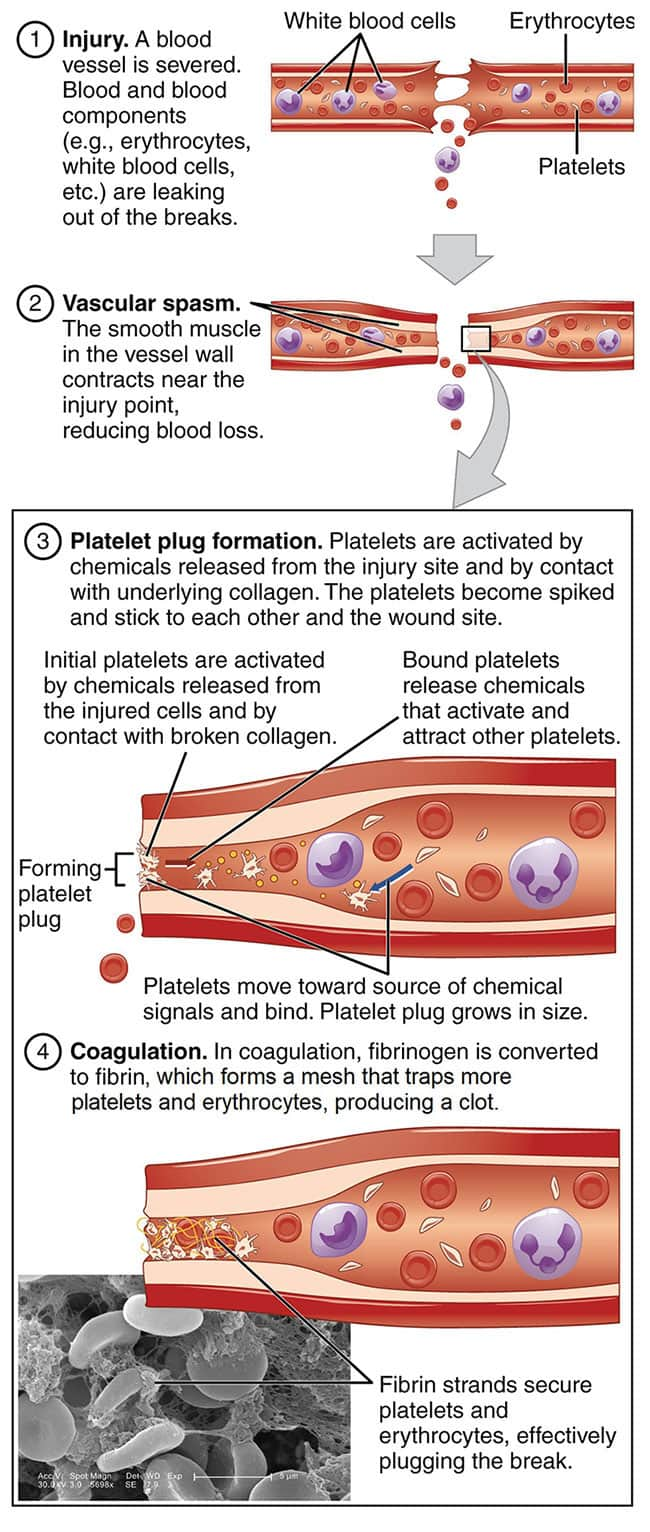
Hemostasis is an intricate process by which blood flow is maintained while both bleeding and excessive blood clotting are avoided. It involves balanced interactions between red and white blood cells, platelets, endothelial cells (the cells that form the inner lining of blood vessels), inflammatory proteins and cytokines, clotting factors, and other circulating proteins.3
Blood clotting is a hemostatic mechanism that prevents blood loss. When a blood vessel injury occurs, the blood vessel constricts and the clotting process, a complex series of overlapping steps, is rapidly set in motion3,11:
- Initiation. Damage to endothelial cells that form the lining of the inner blood vessel exposes an underlying layer of collagen and cells that express an important pro-coagulation protein called tissue factor. The interaction between platelets and tissue factor helps stimulate clot formation by triggering conversion of prothrombin into thrombin, a reaction that requires vitamin K1. The enzyme thrombin activates platelets and stimulates conversion of circulating fibrinogen molecules into strands of fibrin.1,2
-
Amplification.
Injured endothelial cells release inflammatory cytokines that transmit
platelet-activating signals and a platelet-binding protein called von
Willebrand factor. Von Willebrand factor also binds to collagen and
acts as a bridge between platelets and collagen in the blood vessel
wall at the site of injury. In addition, it plays a role in the
clotting cascade—a sequence of reactions that convert clotting factors
into enzymes and ultimately leads to the production of thrombin and
fibrin. Vitamin K1 is a cofactor needed for several steps in the
clotting cascade.2,11
Activation signals cause platelets to change shape and release pro-coagulant proteins, including von Willebrand factor and tissue factor, facilitating their ability to bind to other platelets (aggregation). The accumulation of platelets forms a plug that seals the site of injury and performs the critical function of preventing blood loss. Activated platelets also release pro-inflammatory molecules and amplify the clotting cascade, accelerating the production of thrombin and fibrin.1,2,11 - Propagation. Activated platelets generate large amounts of thrombin. They also bind to fibrinogen, which, when converted to fibrin through the action of thrombin, forms a structural matrix that strengthens and stabilizes the blood clot.
- Fibrinolysis. The blood clot, or thrombus, is broken down during tissue repair through an enzymatic process called fibrinolysis, during which a protein fragment known as D-dimer is released.2,11
In healthy conditions and the absence of blood vessel injury, the tendency to form venous and arterial clots is kept in check by molecules that inhibit activation of the clotting cascade and promote fibrinolysis. These include tissue factor pathway inhibitor, antithrombin, heparin, and proteins C and S (which are also vitamin K-dependent).1 In addition, anti-inflammatory pathways are favored and endothelial cells produce nitric oxide, which helps keep blood vessels dilated and inhibits platelet adhesion.12
4 Thrombosis
Hemostasis is tightly controlled, but conditions that alter regulatory mechanisms can shift the balance in favor of coagulation and increase the risk of excessive blood clotting or abnormal formation of blood clots, known as thrombosis.2,3 Thrombosis can occur as venous or arterial clots, and while venous and arterial thrombosis are distinct pathologies, both are related to inflammation, endothelial cell damage, and platelet activation, and share common risk factors.5
Venous Thrombosis
Blood clots that form in the veins are mainly composed of red blood cells with an outer layer of platelets held together by a protein matrix formed by fibrin.15 Deep vein thrombosis (DVT) is the most serious form of venous thrombosis. Venous stasis (lack of blood movement in a vein) is the most important promoter of DVT, but vascular injury and hypercoagulability (a high propensity to form clots) also contribute to risk.16 DVT usually occurs in the lower legs, but can occur in any deep vein, including the cerebral, abdominal, and retinal veins.17 In about 20% of cases, DVT causes blockage of blood flow away from the tissue, resulting in swelling, pain, and other symptoms depending on its location.17,18
Approximately one-third of DVT patients, especially those with DVT in the legs, experience pulmonary embolism.16 This occurs when a blood clot breaks free and travels through the bloodstream to the lungs, where it can become lodged and obstruct blood flow. Venous thromboembolism (VTE) is a condition that includes DVT and pulmonary embolism and affects approximately 375,000–425,000 people in the United States annually.19 About 10‒30% of DVT and pulmonary embolism episodes cause death within 30 days (with the risk of death being higher in pulmonary embolism), and the 10-year mortality rate among those with venous thromboembolism is reported to be as high as 40%.18,20-22 DVT patients also have a high risk of experiencing recurrences (roughly one-third of cases) and developing long-term complications such as post-thrombotic syndrome and chronic pulmonary hypertension.18 Post-thrombotic syndrome is caused by thickening and scarring of the vein due to DVT-induced vascular inflammation and is marked by venous hypertension. Symptoms include pain, heaviness, swelling, and sometimes ulceration in the limb affected by DVT.19,20
Venous thrombosis can also occur in superficial veins, usually in the legs and frequently due to varicosities. In most cases, superficial vein thrombosis resolves without treatment, but some cases evolve into DVT with a risk of pulmonary embolism.9 In fact, reports suggest as many as 18.1% of superficial vein thrombosis cases involve deep veins and 6.9% result in pulmonary embolism.23 In particular, superficial vein thrombosis that occurs near a critical venous junction in the upper leg carries the same risks as DVT and is treated similarly. A large thrombus (more than 1.5–2 inches in length) in a superficial vein can also lead to complications or post-thrombotic syndrome and may warrant medical management.9
Arterial Thrombosis
Blood clots that form in arteries, or arterial clots, are mainly made of platelets and fibrin, with small numbers of red and white blood cells trapped in the matrix.15 Arterial thrombosis causes reduced blood flow (ischemia), resulting in reduced oxygen and nutrient delivery and ischemic tissue damage. A blood flow-obstructing clot that develops in a coronary artery can lead to acute coronary syndrome (unstable angina or heart attack), and in a cerebral artery, it can cause a stroke.3,12 An arterial blood clot can also break away from the vessel wall. The dislodged or fragmented blood clot may then travel as an embolus, become lodged in a distant artery, and block downstream blood flow, causing acute symptoms.12,24 For example, blood clots that form in the coronary vessels often become embolic (thromboembolism) and travel to the cerebral vascular system, where they can cause transient ischemic attack (TIA) or ischemic stroke.25,26 The aorta and carotid arteries are other frequent sites of thromboembolism.27
One of the major contributors to arterial thrombosis is atherosclerosis. In addition to contributing to altered blood flow, atherosclerotic plaques are associated with vascular dysfunction and chronic inflammatory signaling, which leads to chronic platelet activation.28-30 Furthermore, the inflammatory state induced by atherosclerosis increases plaque vulnerability to destabilization and rupture—an event that triggers pronounced pro-coagulation processes.28,31,32 Another common trigger of arterial thrombosis is atrial fibrillation, a type of arrhythmia. Atrial fibrillation causes turbulence of blood flow through the heart chambers and is associated with a five-fold increase in embolic stroke risk.33
5 Dietary and Lifestyle Considerations
Dietary and lifestyle interventions are crucial components of a program to reduce thrombosis risk. Habits that reduce oxidative stress, inflammatory signaling, LDL-cholesterol levels, and blood pressure, support metabolic health and weight maintenance, and promote gut microbial balance are key to preventing both venous and arterial clots and their consequences (eg, VTE or post-thrombotic syndrome).34
Dietary patterns high in plant foods can supply ample amounts of antioxidant nutrients, mono- and polyunsaturated fatty acids, vitamins, minerals, fiber, and phytochemicals that are associated with anti-inflammatory, platelet activation-inhibiting, and antithrombotic effects.35-37 A Mediterranean diet in particular is well studied for its positive impacts on cardiovascular and metabolic health and conditions associated with thrombosis risk.35,36 Foods that characterize a Mediterranean diet, such as olive oil, fruits and vegetables, nuts and seeds, whole grains, legumes, and fish, have each been found to reduce venous and arterial thrombosis risk in observational and animal studies.38 Dietary Approaches to Stop Hypertension (DASH) is another set of dietary guidelines that promotes a mainly plant-based diet and, like a Mediterranean diet, is linked to broad health benefits that could potentially reduce excessive blood clotting and the risk of thrombosis.36
Blood Thinning Foods, Drinks & Nutrients
Some specific foods have been found to lower the risk of thrombosis, including:
- Dark chocolate. Cocoa contains flavanols that have demonstrated antithrombotic effects.39,40 Consumption of dark chocolate has been linked to cardiovascular health and lower risk of conditions related to thrombosis like hypertension, coronary and peripheral vascular disease, and atrial fibrillation.41 One clinical trial reported 300 mL of a flavonol-rich chocolate beverage had a similar, though weaker, effect on platelets to 80 mg of aspirin in healthy adults.42
- Onion. Laboratory and animal research showed onion extracts reduced platelet activation and thrombosis. This may be due to onions’ high content of the flavonoid quercetin.43 An observational study noted individuals with higher consumption of onions and garlic had a lower incidence of cardiovascular events.44 In a crossover trial, blood samples taken after eating a bowl of onion soup or a control soup suggested onions may have an immediate inhibiting effect on platelet aggregation.45
- Garlic. Garlic and its active constituents have been shown to reduce inflammation, scavenge free radicals, and improve lipid metabolism.46 Garlic and onion intake has been associated with reduced cardiovascular risk.44 A clinical trial found eating one clove of fresh garlic per day for 16 weeks lowered levels of an enzyme that promotes constriction of blood vessels and stimulates platelet aggregation.47
- Ginger. Preclinical evidence suggests ginger can inhibit platelet aggregation, lower blood pressure, reduce inflammatory cytokine levels, and improve lipid levels,48,49 although findings from clinical trials have been mixed.50 An observational study found increased ginger intake was correlated with lower risk of coronary artery disease in older adults.51
- Tomato. Tomatoes are rich in lycopene and other oxidative stress-reducing compounds that have demonstrated antithrombotic effects.52 Cooking tomatoes with oil enhances lycopene bioavailability.53 A study that included almost 24,000 participants found higher tomato intake was correlated with better markers of cardiovascular health and lower risk of death due to coronary artery disease or stroke.54 A meta-analysis of 25 observational studies found the highest consumption or serum levels of lycopene were associated with the lowest incidences of cardiovascular disease and stroke.55
- Fruit. Citrus fruit and citrus juice have demonstrated platelet inhibition in laboratory studies.56 In clinical studies, purple grape juice lowered platelet reactivity in healthy subjects.57
- Fish. Fish provides anti-inflammatory omega-3 fatty acids, and regular intake has been found to reduce platelet aggregation in human studies.58 In one observational study that included 2,033 men who recently experienced a heart attack, the addition of two fish servings per week was associated with 29% lower mortality risk over two years of monitoring.59 Higher dietary intake of fish fatty acids was also associated with lower risk of recurrent VTE in a study in patients with a previous DVT and without cancer or another known trigger of the first event, and the relationship was stronger in those not using anticoagulant (blood thinner) drugs.60 Some evidence suggests eating a diet with a lower omega-6:omega-3 fatty acid ratio may reduce thrombosis risk. Although the exact optimal ratio of omega-6:omega-3 fatty acid intake remains in question, it is increasingly clear that lower ratios, such as less than 4:1, are associated with lower risk of heart disease and other chronic conditions.61,62
- Olive oil. Extra virgin olive oil (EVOO) is rich in monounsaturated fatty acids and antioxidant polyphenols, and high intake has consistently been associated with lower cardiovascular risk.63 Evidence suggests olive oil may suppress the rise in coagulation enzyme activity typically induced by a high-fat meal.64 Including 30 mL (one ounce) of olive oil in the daily diet for four months was found to improve vascular function and lower inflammation in individuals with early signs of atherosclerosis.65
- Alcohol. Light-to-moderate alcohol consumption, especially red wine, has been correlated with cardiovascular benefits, including reduced risk of thromboembolic events and deaths.66-68 Light-to-moderate drinking consists of a maximum of two drinks per day for men and one drink per day for women, with a drink being 12 ounces of beer, 5 ounces of wine, or 1.5 ounces of spirits.69 However, heavy consumption is linked to poor cardiovascular health, increased risk of atrial fibrillation, and higher rates of cardiovascular events.68,70 The relationship between alcohol consumption and venous thrombosis is uncertain.71-74
It is important to note that many foods, particularly those rich in vitamin K, can interact with warfarin. Among the foods that can interfere with warfarin’s action (increasing the likelihood of excessive blood clotting) are green leafy vegetables (eg, kale, spinach, collards, Swiss chard, broccoli, Brussels sprouts, cabbage, lettuce), green tea, soy foods, blueberries, and soybean and canola oils. On the other hand, grapefruit, cranberry juice, mango, and alcohol are examples of foods that may increase bleeding risk in patients taking warfarin by slowing its metabolism.75,76 Evidence from observational and experimental research suggests dietary stability is more important than dietary restriction during warfarin therapy.77 Warfarin-treated patients should consult with their healthcare provider before making dietary changes.
Exercise
A sedentary lifestyle is a risk factor for arterial thrombosis and individuals who engage in regular exercise appear to be less susceptible to thrombosis.78,79 Overall, moderate-intensity exercise can improve vascular function, reduce oxidative stress, lower levels of proteins involved in clotting, and reverse conditions that lead to pro-thrombotic signaling.79 Regular exercise may even help regulate heart rhythm and lower the risk of atrial fibrillation.80 In a randomized controlled trial, the addition of high-intensity interval training to a 12-week moderate-intensity exercise program reduced platelet reactivity and aggregation compared with moderate-intensity exercise alone.81 On the other hand, a bout of high intensity physical exertion can sometimes trigger a thromboembolic event, particularly in untrained individuals, possibly due to vascular injury and higher levels of stress hormones caused by acute exercise.78,79
Although evidence suggests aerobic exercise may also decrease the likelihood of VTE, the protective effect does not appear to be as strong as for arterial clots.82 Nevertheless, controlled trials have found targeted exercises, such as ankle, lower limb, or hand exercises, can reduce the risk of venous thrombosis related to surgery or catheter placement.83-85
Stress Management
Stress is a major driver of inflammation and coagulation and the conditions associated with thrombosis.29,86 Acute stress, negative emotions, and psychological trauma can precipitate thromboembolic events by triggering atherosclerotic plaque rupture. Individuals with poor vascular health are especially vulnerable to heart attack and stroke within two hours after an intense psychological stressor.87 In addition, chronic stress has been found to more than double the risk of heart attack.29 Both acute and chronic stress signaling via hormones and neurotransmitters can increase thrombosis risk by increasing platelet numbers, reactivity, and aggregation, disrupting normal vascular function, up-regulating the clotting cascade, and altering fibrinolysis mechanisms. The strongest effects of stress are seen in those with pre-existing cardiovascular disease.86
Meditation and stress management practices may lower the risk of thrombosis. In a study that included data from more than 61,000 participants, those who reported having any meditation practice were noted to have a lower incidence of heart attack and stroke.88 In a randomized controlled trial in 47 patients referred to cardiac rehabilitation after a heart attack or coronary procedure, those who received eight weeks of mindfulness-based stress reduction training had greater improvement in cardiovascular health markers, as well as lower depression and anxiety scores and more improved health-related quality of life, than those who received standard care.89
6 Nutrients
There are many nutritional options for potentially lowering the risk of blood clots or excessive blood clotting. If you are taking an antiplatelet or anticoagulant (blood thinner) medication, it is important to note there may be additive effects from these supplements, which could lead to increased bleeding risk. There is also a risk of drug‒supplement interactions that could reduce the effectiveness of anticoagulant medications, especially warfarin. If you are taking any anticoagulant or antiplatelet medications, a consultation with the prescribing health care provider is essential before adding any of these supplements to your program.
The following nutrient supplements have evidence supporting their possible ability to promote balanced hemostasis and reduce blood clot risk. A comprehensive approach to preventing blood clots includes maintaining optimal cardiovascular and metabolic health. You can find more guidance in the Atherosclerosis and Cardiovascular Disease protocol, as well as protocols for other associated conditions.
Fish Oil
Fish oil-derived omega-3 fatty acids, particularly eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), are used by cells to make compounds that affect hemostasis and thrombosis by modulating platelet and endothelial function. EPA and DHA have been shown to reduce platelet reactivity and aggregation, increase production of nitric oxide, inhibit oxidized LDL generation, and decrease expression of adhesion molecules on endothelial cell surfaces. They may also promote fibrinolysis and inhibit the coagulation cascade by reducing tissue factor expression.61
A meta-analysis of data from eight randomized controlled trials found a combination of omega-3 fatty acids plus statin therapy was more effective than statin therapy alone for stabilizing and promoting regression of atherosclerotic plaques in the coronary arteries, possibly reducing the risk of arterial thromboembolism.90 Another meta-analysis of 40 randomized controlled trials with a combined total of 135,267 participants found supplementing with EPA and DHA lowered the risk of heart attack and cardiovascular death. The analysis further calculated that the risk of cardiovascular events was reduced by 5.8% for every additional gram of EPA plus DHA taken daily.91
A study performed using observational evidence from three large cohort studies indicated higher blood levels of DHA were associated with lower risk of atherothrombotic stroke (caused by arterial clots formed in a cerebral artery) and higher blood levels of docosapentaenoic acid (DPA) were associated with lower risk of cardioembolic stroke (cause by an embolism from a coronary artery).92 DPA is another omega-3 fatty acid found in fish; it can be made in the body from EPA and further converted into DHA.
However, a meta-analysis of 14 trials in 125,763 subjects found, despite their association with lower risk of heart attack and other major cardiovascular events, taking 1 gram or more of omega-3 fatty acids was associated with increased risk of bleeding and of developing atrial fibrillation.93
It is important to note that the results of multiple meta-analyses showing an increased risk of atrial fibrillation with omega-3 fatty acids are driven by studies that have methodological characteristics that limit their extrapolation to typical fish oil supplements. For example, some of the studies in these meta-analyses utilized high dosages of EPA-based pharmaceutical preparations (eg, icosapent ethyl [Vascepa, among others]). Also, in an attempt to improve the pharmacological effects of omega-3 fats, some of the studies used fatty acid forms (eg, carboxylic acid-based EPA and DHA preparations) other than the ethyl ester form that has been extensively studied.94 A large ongoing cohort study of a multi-ethnic population suggests both EPA and DHA play important roles in cardiovascular health. Preliminary analysis of data from this study highlights an association between EPA levels and reduced bleeding risk as well as DHA levels and reduced atrial fibrillation risk. These observations draw attention to the role that DHA may play in reducing atrial fibrillation as well as the overall cardiovascular benefits that have been reported with fatty fish and omega-3 intake.95 Preclinical research also points toward the importance of DHA in reducing atrial fibrillation risk.96 However, since the totality of the literature is inconsistent and complex, caution is advised for individuals with a history or high risk of atrial fibrillation. More information on this topic can be found in Life Extension’s Arrhythmias protocol.
A randomized placebo-controlled trial in 452 elderly participants requiring leg surgery due to a fracture found 1 gram of omega-3 fatty acids, taken for 30 days following surgery, reduced the risks of DVT and pulmonary embolism without increasing bleeding events or other complications.97 In 567 patients with chronic kidney disease, 4 grams of omega-3 fatty acids daily for three months reduced the risk of acute thrombosis in the arterio-venous fistulas used for dialysis.98 An observational study with 21,970 participants found a weekly intake of more than 4.7 grams of fish omega-3 fatty acids from diet and supplements was correlated with a 22–26% lower risk of VTE and 39–60% lower risk of pulmonary embolism after a median follow-up period of 11.6 years. 99 Higher omega-3 fatty acid intake has also been associated with lower rate of recurrence of DVT or other venous thromboembolic events, especially in those whose first event had no known trigger (eg, surgery, hospitalization, cancer, or acute illness).60,100
Ginkgo
Ginkgo (Ginkgo biloba) has been used for centuries as an anti-aging medicine. Ginkgo extract has been found to stimulate peripheral and coronary circulation and may protect against neurological and cardiovascular disorders.101-104
Studies show compounds from ginkgo reduce oxidative and inflammatory endothelial cell injury and improve vascular function, inhibiting atherosclerotic plaque development and possibly decreasing the risk of arterial clots.105,106 In a clinical trial involving patients at high risk of venous thrombosis due to chronic venous insufficiency, treatment with ginkgo extract led to reduced levels of circulating endothelial cells. Circulating endothelial cells are considered a marker of ongoing vascular injury.107 Evidence also suggests ginkgo may improve conditions associated with thrombosis, including high blood pressure, unhealthy lipid levels, and high blood glucose levels.108
Ginkgo extract has been shown in preclinical studies to inhibit platelet activating factor (involved in platelet activation) and increase endothelial cell expression of thrombomodulin (activates thrombin) and production of tissue plasminogen activator (stimulates fibrinolysis).109,110 Other research shows ginkgo flavonoids can directly inhibit thrombin activity.111 However, ginkgo does not appear to affect bleeding time and its use has not been associated with an increased risk of bleeding.112,113
Garlic
A growing body of research indicates garlic can slow progression of atherosclerosis and improve cardiometabolic health.46 Garlic and garlic extracts may lower thrombosis risk by inhibiting platelet aggregation, reducing fibrinogen levels, and increasing fibrinolytic processes.114,115 In animal research, garlic delayed platelet aggregation and clot formation in response to thrombotic triggers.116,117 Some evidence suggests ajoene, an active compound from garlic, may contribute to its antithrombotic properties.118,119
In a clinical trial, 36 healthy young men received 600 mg, 1,200 mg, or 2,400 mg of garlic extract or 75 mg of the blood thinner drug clopidogrel daily for three weeks. Blood samples from those who received 1,200 mg or 2,400 mg of garlic showed significantly reduced platelet aggregation in response to various triggers, and a stronger effect was seen with the higher dose. The 2,400 mg dose of garlic was more effective than clopidogrel for inhibiting platelet response to some but not all thrombotic triggers. In addition, bleeding time was prolonged (but stayed within the normal range) in all participants, with the longest bleeding time seen in those taking 2,400 mg of garlic extract.120 A 12-week placebo-controlled trial examined the safety of combining aged garlic extract with warfarin therapy. The trial included 48 closely monitored participants and found taking aged garlic extract along with warfarin did not increase bleeding or other adverse side effects.121
Cocoa
Cocoa is rich in flavanols and other polyphenols with strong free radical-scavenging effects. Cocoa and dark chocolate have been found to reduce platelet activation, inhibit expression of proteins involved in platelet aggregation, and limit the effects of platelet activity on hemostasis.39,40 In addition to its polyphenols, cocoa is a source of theobromine, a compound in the methylxanthine family, that also inhibits platelet aggregation.40 Clinical trials have found polyphenols from cocoa increase nitric oxide production and support healthy endothelial function, decrease inflammatory marker levels, reduce oxidative damage to lipids, and promote growth of beneficial bacteria in the gut.40,122,123
In clinical trials in healthy men, eating 50 grams (1.67 ounces) of chocolate with 90% cocoa led to reduced platelet reactivity and prolonged clotting time in blood samples taken four hours later.124,125 Another clinical trial found dark chocolate consumption blunted the rise in D-dimer levels caused by an experimental psychological stressor in healthy men.126 In an open-label controlled trial in healthy middle-aged adults, 900 mg of cocoa flavanols per day for 30 days improved endothelial function, an important factor in thrombotic risk, and in a follow-up controlled trial the same dose was also shown to improve blood pressure and lipid levels.127 A randomized controlled trial found cocoa providing 750 mg of flavanols per day improved vascular function and markers of endothelial integrity more than low-flavanol cocoa after 30 days in subjects with coronary artery disease.128 Furthermore, flavanol-rich cocoa has also been found to improve vascular function in young African Americans,129 healthy young adults,130 post-menopausal women,131 patients with end-stage kidney disease,132 men with high blood pressure,133 and healthy elderly men.134
Coenzyme Q10
Coenzyme Q10 (CoQ10), which may be in the reduced form as ubiquinol or non-reduced form as ubiquinone, is a nutrient involved in reduction-oxidation balance and energy production in the mitochondria. A low CoQ10 level increases inflammatory signaling and is linked to poor cardiovascular health and outcomes, and CoQ10 supplements have a potential role in preventing and as an adjuvant to treating cardiovascular disease and associated metabolic conditions.135-138 CoQ10 has been found to improve vascular function by reducing oxidized LDL accumulation in arterial walls, decreasing vascular injury and stiffness, and increasing nitric oxide availability.137 Laboratory research further suggests CoQ10 may inhibit platelet aggregation and signaling, reducing thrombus growth.139 Because the use of a common class of cholesterol-lowering drugs called statins lowers CoQ10 production, patients taking statins in particular may benefit from supplementing with CoQ10.136,138,140
In a randomized controlled trial in 213 elderly individuals with low selenium status, taking 200 mg CoQ10 plus 200 mcg selenium for four years led to reduced D-dimer levels. D-dimer levels are a reflection of thrombotic potential. In participants whose baseline D-dimer levels were higher than the median (mid-point), those given CoQ10 plus selenium had a lower incidence of cardiovascular death during 4.9 years of follow-up.141 The same combination of CoQ10 and selenium was also found to reduce levels of von Willebrand factor and tissue plasminogen activator inhibitor-1, molecules with critical roles in promoting thrombosis, in another four-year placebo-controlled trial in 308 elderly individuals with low selenium status.142 In a randomized controlled trial in 51 subjects with high LDL-cholesterol levels, supplementing with 100 or 200 mg of CoQ10 daily for eight weeks led to improved endothelial function, reduced LDL oxidation, and increased nitric oxide availability compared with placebo.143 In a randomized placebo-controlled crossover trial in 36 patients with antiphospholipid syndrome, an immune disorder associated with an increased risk of thrombosis, 200 mg CoQ10 daily for one month improved vascular function; lowered markers of thrombotic risk, inflammation, and oxidative stress; and modified expression of 23 of 29 atherosclerosis-related genes.144
French Maritime Pine Bark Extract (Pycnogenol)
Pycnogenol, an extract from French maritime pine bark, has been reported to prevent edema and venous thrombosis during long airplane flights and may help chronic venous insufficiency.145,146 In one controlled trial, 198 airline passengers with moderate to high risk of venous thrombosis were given either pycnogenol, at 200 mg before and during a long flight (averaging 8 hours 15 minutes) plus 100 mg the next day, or placebo. There were five venous thrombotic events in the placebo group and none in the pycnogenol group.147
In a randomized controlled trial in 156 subjects with a history of a single DVT episode, 150 mg pycnogenol was as effective as compression stockings at reducing edema after one year, but was associated with better compliance due to the discomfort of compression stockings.148 In an observational study that followed 222 subjects with a history of DVT for six years, those taking 200 mg pycnogenol per day had lower risks of recurrent DVT and post-thrombotic syndrome compared with those treated with blood thinner medications such as aspirin, ticlopidine (Ticlid), sulodexide (Aterina, a formulation composed of 80% low-molecular weight heparin), or no medication.149 Pycnogenol has also been found to improve arterial endothelial function by increasing nitric oxide production in healthy subjects and those with coronary artery disease.150,151 One research team found a dose of 200 mg pycnogenol per day for two months suppressed the smoking-induced rise in platelet aggregation in chronic smokers but had no effect on platelet aggregation in non-smokers.152
Observational evidence from a study in 307 subjects with a history of retinal vein thrombosis found taking 100 mg pycnogenol daily was associated with the lowest incidence of repeat retinal vein thrombosis compared with aspirin, ticlopidine, sulodexide, or no medication, and was the only treatment associated with reduction in edema.153 A smaller study of similar design found pycnogenol, at 100 mg daily, was associated with better vision, lower risks of retinal edema and recurrent retinal vein thrombosis, and lower frequency of adverse side effects compared with aspirin after nine months.154
Probiotics
The relationship between the gut microbiome and cardiovascular health is an area of intense research. A growing body of evidence suggests dysbiosis (an imbalance in microbiome composition) is an important underlying contributor to atherosclerosis and thrombosis.155,156
A meta-analysis of 15 randomized controlled trials with a combined total of 976 participants found supplementing with Lactobacillus species, especially L. reuteri and L. plantarum, can lower total and LDL-cholesterol levels, potentially reducing cardiovascular risk.157 The probiotic strain L. reuteri NCIMB 30242 has demonstrated several positive effects that might lower thrombosis risk. In a randomized placebo-controlled trial in 127 subjects with high cholesterol levels, taking a supplement providing at least 4 billion colony forming units (CFUs) of L. reuteri NCIMB 30242 per day for nine weeks reduced LDL-cholesterol levels, as well as hs-CRP, apolipoprotein B-100 (apoB-100) (a lipid fraction associated with atherosclerosis development), and fibrinogen levels, and increased vitamin D levels.158,159 Similarly, a yogurt providing 1.4 billion CFUs of L. reuteri NCIMB 30242 taken twice daily for six weeks reduced LDL-cholesterol, apoB-100, and non-HDL-cholesterol levels compared with placebo yogurt in a trial that included 114 individuals with high cholesterol levels.160
Other probiotic strains have been found to improve parameters related to thrombosis risk. In a placebo-controlled trial in 34 healthy individuals, consuming yogurt fortified with 10 billion CFUs of Bifidobacterium animalis subspecies lactis plus 600 mg of the amino acid arginine (a precursor to nitric oxide) daily for 12 weeks improved vascular function and was therefore suggested to reduce atherosclerosis risk.161 A placebo-controlled trial in 36 heavy smokers found a drink providing 20 billion CFUs of L. plantarum per day for six weeks decreased blood pressure and fibrinogen levels, as well as levels of an inflammatory cytokine and a marker of oxidative stress.162 Another trial in 32 patients with human immunodeficiency virus (HIV) compared the effect of a daily multi-strain probiotic supplement to placebo or no supplement; those who received the probiotic had greater reductions in D-dimer levels after eight weeks.163 In mice, intake of a milk product fermented with a Bacillus subtilis strain that produces the thrombolytic enzyme nattokinase reduced the development of thrombosis.164
Nattokinase
Nattokinase is a thrombolytic enzyme produced by certain bacteria and found in a fermented soybean product called natto. Through its enzymatic action and ability to activate plasmin, nattokinase breaks down the fibrin structure that holds blood clots together.165 It has also been shown to decrease levels of fibrinogen, factor VII, and factor VIII in multiple subgroups of participants when taken at a dose of 2,000 fibrinolytic units daily for two months.166 In a controlled trial in 204 individuals at high risk of venous thrombosis, a combination of French maritime pine bark (Pycnogenol) plus nattokinase or placebo was taken before and after a seven- to eight-hour airplane flight; no venous thrombi occurred in the pycnogenol/nattokinase group, while five DVT and two superficial venous thrombi occurred in the placebo group. In addition, pycnogenol plus nattokinase-treated subjects had a 15% reduction in edema at the end of the flight versus a 12% increase in edema in the placebo group.167
Nattokinase has been found in animal studies to prevent arterial thrombosis and dissolve existing blood clots.168 Evidence suggests nattokinase reduces the formation of intravascular blood clots by reducing oxidative stress and inflammatory signaling.169-171 Nattokinase has also been found to reduce thickening of the femoral artery wall in response to arterial injury in research animals.172,173 Unfortunately, in a randomized placebo-controlled trial in 265 healthy elderly subjects, no difference in carotid artery wall thickness (an indicator of atherosclerosis), measured using ultrasound imaging, were seen in those who received 2,000 fibrinolytic units of oral nattokinase per day versus placebo for three years.174
B Vitamins
Niacin (vitamin B3) is one of the more effective agents for raising high-density lipoprotein (HDL)-cholesterol levels.175 HDL has antithrombotic effects due to its ability to protect endothelial cells from oxidative and other injury and to remove excess cholesterol from the artery wall, preventing plaque formation that can lead to thrombosis.176 Before the development of statin drugs, niacin was commonly used to treat abnormal lipid profiles. Because recent evidence shows little benefit on cardiovascular outcomes from adding extended-release niacin to statin therapy, or from raising HDL-cholesterol levels, niacin is no longer considered standard of care for reducing cardiovascular risk.175-178 Nevertheless, niacin has demonstrated antithrombotic effects, including inhibiting a coagulation cascade enzyme called factor VII, reducing fibrinogen levels, decreasing expression of tissue factor, suppressing plasminogen activator inhibitor-1 (an enzyme that inhibits fibrinolysis), lowering blood viscosity, and decreasing platelet aggregation.179
Vitamins B6, B12, and folate are needed for metabolism of homocysteine, high levels of which are associated with vascular damage and increased thrombosis risk. Low levels of these B vitamins, especially folate and B12, and high homocysteine levels are associated with increased risk of atherosclerosis, stroke, and venous thrombosis.180-182 For example, in patients with atrial fibrillation, high homocysteine (often from B12 deficiency) increases the risk of stroke four-fold.183 In addition, pernicious anemia, a condition that impairs intestinal B12 absorption, has a well-documented association with high homocysteine levels and elevated risk of venous thrombosis.184,185
Lowering homocysteine levels through supplementation with folic acid and B12 has been shown to lower the risk of stroke.183,186 In a randomized controlled trial, 10,789 Chinese adults with high blood pressure were treated daily either with enalapril (Vasotec) (a blood pressure-lowering drug) or enalapril along with 800 mcg folic acid and followed for 4.2 years. Subjects with low platelet numbers and high homocysteine levels had the highest stroke risk, and in this high-risk subgroup, the risk was 73% lower in those receiving folic acid.187 In another controlled trial in stroke patients with a history of DVT, lowering high homocysteine levels using folic acid and B12 was shown to reduce the rate of DVT recurrence from 28.9% to 4.4%.188
Green Tea
Green tea contains bioactive polyphenols known as catechins, the most studied of which is epigallocatechin gallate (EGCG). Clinical trials and observational studies have found green tea is associated with reduced cardiovascular risk due to increased nitric oxide availability, reduced inflammation and oxidative stress levels, and improved endothelial function.189,190 Research using blood samples from subjects given the blood thinner medications aspirin, clopidogrel, or ticagrelor (Brilinta) suggests EGCG may improve their antiplatelet effects without increasing the risk of bleeding.191 EGCG has been shown to inhibit inflammatory enzyme activity in platelets192 and suppress inflammatory signaling, inhibit expression of adhesion molecules, and promote expression of nuclear factor erythroid 2-related factor 2 (Nrf2, a protein that upregulates antioxidant pathways) in endothelial cells.189,193 EGCG also inhibited expression of tissue factor, a protein that triggers coagulation, on arterial and venous endothelial cells.194 In laboratory animals, EGCG inhibited platelet aggregation, prevented thrombus formation in injured vessels, and prolonged bleeding time.194,195
Lycopene
Lycopene is a carotenoid found in high concentrations in tomato skins. Laboratory and animal research suggests lycopene may suppress tissue factor activity and inhibit platelet aggregation.196-198 Lycopene has been suggested to potentially prevent thrombosis through mechanisms such as reducing endothelial injury, inhibiting oxidation of LDL, reducing inflammatory immune activity, and decreasing cholesterol synthesis.199 A meta-analysis of 28 observational studies found higher intake or blood levels of lycopene were correlated with a lower risk of stroke and cardiovascular disease.55
Tomato pomace is a byproduct of tomato product manufacturing and contains mainly tomato seeds and skins. Tomato pomace contains lycopene and a number of tomato flavonoids that may work together to inhibit platelet aggregation and reduce thrombosis risk.200,201 In a placebo-controlled trial in 99 healthy young men, taking 1 gram of tomato pomace extract daily for five days reduced platelet aggregation.202
Olive Oil and Olive Leaf
Extra virgin olive oil (EVOO) is high in monounsaturated fatty acids and antioxidant polyphenols, and high intake has consistently been associated with lower cardiovascular risk.63 Olive oil has been shown to lower oxidative stress and inflammatory marker levels, improve endothelial function and lipid and carbohydrate metabolism, and inhibit thrombosis.63,203 Preclinical research shows olive oil can reduce platelet activity and aggregation, reduce endothelial cell expression of adhesion molecules, and decrease the coagulation enzyme factor VII and plasminogen activator inhibitor-1, both of which are implicated in coronary artery disease.204 Some evidence suggests olive oil may mitigate the rise in coagulation enzyme activity typically induced by a high-fat meal.64 In one clinical trial in 82 patients with early atherosclerosis, 30 mL (one ounce) of olive oil per day for four months improved vascular function and inflammatory marker levels.65
Oleuropein and its derivative hydroxytyrosol are polyphenols found in olives, EVOO, and olive leaf. These compounds have demonstrated inflammation- and oxidative stress-lowering properties, and have been found to improve vascular function as well as glucose and lipid metabolism.205 Findings from multiple preclinical studies suggest oleuropein, hydroxytyrosol, and other phenolics extracted from EVOO and olive leaf can inhibit platelet activation and aggregation.206-209
Pomegranate
Pomegranate is rich in flavonoids, tannins, and other polyphenols that have strong free radical-scavenging and anti-inflammatory properties.210
In laboratory studies, pomegranate extracts have been found to reduce platelet activation and aggregation in response to thrombotic triggers.211,212 A meta-analysis of eight randomized controlled trials found drinking up to 8 ounces of pomegranate juice per day reduced high systolic blood pressure and more than eight ounces per day reduced both systolic and diastolic blood pressure.213 In an open trial in 13 healthy subjects, drinking 50 mL (almost two ounces) of pomegranate juice per day for two weeks reduced platelet aggregation by 11%.214 Another clinical trial found subjects who drank pomegranate juice exhibited prolonged clotting times six hours later.215
Pomegranate has also been found to reduce oxidative stress and high blood pressure, and improve vascular function and glucose and lipid metabolism.210,216,217 A randomized placebo-controlled trial in 100 heart disease patients found 450 mg of pomegranate extract plus 180 mg vitamin E (as synthetic dl-alpha tocopherol) daily for eight weeks reduced expression of two vascular adhesion proteins and lowered levels of inflammatory markers.218 In another placebo-controlled trial in 48 subjects who were overweight or obese, 1,000 mg pomegranate extract for 30 days reduced inflammatory marker levels and improved blood glucose, insulin, and LDL-cholesterol levels.219
Yerba Mate
Yerba mate (Ilex paraguariensis) is high in chlorogenic acids that have anti-inflammatory and free radical-scavenging properties.220 In a randomized placebo-controlled trial in 142 participants with high blood viscosity, a condition related to increased thrombosis risk, 5 grams of yerba mate tea per day for six weeks led to reduced blood viscosity and improved circulation in small vessels.221 Another controlled clinical trial found 580 mg of chlorogenic acids from yerba mate per day improved markers of cardiometabolic health in 34 middle-aged men at high risk of metabolic syndrome.222 In preclinical research, a constituent from yerba mate reduced thrombin activation and venous thrombosis.223
Some evidence suggests yerba mate tea, when consumed in large amounts and especially when consumed very hot, may raise the risk of esophageal and head and neck cancers. It is thought this possible carcinogenic effect may be related to the presence of polycyclic aromatic hydrocarbons produced through a drying procedure involving smoke. Although a carcinogenic effect for yerba mate tea has not been confirmed, it is prudent to drink yerba mate tea at a slightly lower temperature and limit intake to less than one liter per day.220,224
Quercetin
Quercetin is a flavonoid found in most fruits and vegetables, with high amounts found in onions, apples, tea, and wine. Higher intake of flavonoids, including quercetin, is associated with lower cardiovascular risk.225 Quercetin supplementation, at doses of 150 mg and 300 mg, has been found to inhibit platelet activation, signaling, and aggregation within 30 minutes in healthy adults.226 Supplementation with 150 mg quercetin for six weeks has also been shown to significantly reduce systolic blood pressure and oxidized LDL levels.227 Preclinical research has shown quercetin reduces expression of adhesion molecules on endothelial cells and platelets, lowers oxidative stress, inhibits LDL oxidation, supports nitric oxide production and healthy blood vessel function, improves glucose metabolism, and reduces inflammatory marker levels.225,228 Quercetin, along with other flavonoids, is thought to be responsible for antithrombotic and cardioprotective properties of the bee products propolis and honey.229,230
Saffron
Saffron’s distinctive red-orange color is due to its carotenoids, including crocin and crocetin. Saffron extracts have been found to suppress platelet aggregation, scavenge free radicals, lower oxidative stress, reduce LDL oxidation, inhibit expression of endothelial adhesion molecules, and improve endothelial function.231,232 Crocetin alone has also demonstrated antithrombotic effects in laboratory and animal studies.233-235 In a randomized controlled trial in 84 subjects with coronary artery disease, 30 mg per day of crocin for eight weeks lowered levels of oxidized LDL and an inflammatory marker (monocyte chemoattractant protein-1, or MCP-1), and inhibited expression of genes related to atherosclerosis compared with placebo.236
Ginger
Ginger is a culinary spice and a medicinal herb that has clinically demonstrated cardioprotective effects such as improving lipid levels, reducing levels of inflammatory cytokines, lowering blood pressure, and inhibiting platelet aggregation.48,49 An observational study that examined health records and dietary surveys from 4,628 participants found, among those 60 years and older, those who reported higher ginger use had a lower incidence of coronary artery disease.51 Ginger may also be beneficial in metabolic disorders such as obesity and type 2 diabetes.237 In a study in mice, zingerone (an active compound from ginger) inhibited not only platelet aggregation but also the activity of factor Xa, an enzyme in the clotting cascade.238 Laboratory research also found a polysaccharide extracted from ginger inhibited coagulation pathways.239 A systematic review of eight clinical trials was unable to draw a firm conclusion about the effect of ginger on platelet aggregation in humans due to the small sample sizes and variable methodologies used.50 In one of the trials, taking 4 grams of ginger for three months did not change platelet aggregation, fibrinogen level, or fibrinolytic activity in subjects with coronary artery disease, but a single dose of 10 grams reduced platelet aggregation.240
Vitamin C
Vitamin C is a water-soluble antioxidant and has demonstrated an ability to reduce platelet aggregation and promote fibrinolysis, without impacting coagulation pathways, in high-oxidative stress pro-thrombotic conditions in the laboratory.241-243 In particular, vitamin C appears to reduce expression of adhesion proteins by endothelial cells and platelets.242,244 A large population study of more than 20,000 adults ranging from 40‒79 years of age followed the individuals for an average of 9.5 years and found that those in the top quartile of baseline plasma vitamin C concentration had a 42% lower risk of stroke than those in the bottom quartile.245 A meta-analysis of 17 randomized controlled trials found vitamin C supplementation at doses of 500 to 2,000 mg per day improved measures of vascular function.246,247 Some evidence suggests vitamin C supplementation may improve atherosclerosis and other conditions associated with increased risk of venous and arterial clots, but the effect is likely to be mild and of greater importance in those with lower vitamin C levels and higher cardiovascular risk.246,247 Indeed, positive effects on blood vessel dilation were seen in patients with chronic heart failure after taking 1 gram of vitamin C twice daily for four weeks.248 In smokers, whom have been proven in multiple studies to have low blood levels of vitamin C, oral supplementation with 2 grams vitamin C per day has been shown to significantly decrease urinary isoprostanes,249 markers of oxidative stress, and monocyte adhesion to the endothelial lining of blood vessels, one of the early stages in the development of dangerous atherosclerotic plaque.250
Grape Seed Extract
Grape seeds are high in polyphenols with health benefits based on their anti-inflammatory, oxidative stress-reducing, and cardiovascular protectant effects. Preclinical research has shown grape seed extract has anticoagulant and antiplatelet properties.251 In mice, grape seed extract reduced platelet aggregation without increasing bleeding.252 Grape seed extract also reduced thrombus formation and spread in a rat model of DVT.253 A meta-analysis of 16 clinical trials found grape seed extract supplementation significantly decreased systolic blood pressure in younger (<50 years) or obese subjects and individuals with metabolic syndrome.254
Capsaicin
Capsaicin is an active compound found in red chili pepper and responsible for their characteristic spicy taste. Capsaicin is used to treat inflammatory, metabolic, and infectious disorders.255 Preclinical and clinical evidence suggests capsaicin can improve lipid and glucose metabolism, support weight loss, and lower the risk of both type 2 diabetes and cardiovascular disease.255-257 Laboratory evidence shows capsaicin has the potential to reduce platelet aggregation, without affecting coagulation, in response to an array of pro-thrombotic triggers, possibly by inhibiting inflammatory pathways.257-261 However, in healthy male volunteers, neither 400 mcg nor 800 mcg capsaicin affected platelet aggregation.262
Resveratrol
Resveratrol is a flavonoid found in purple grapes, red wine, peanuts, soybeans, and berries. It has well-established anti-inflammatory and free radical-scavenging effects and may slow the effects of aging on blood vessels.263-265 Preclinical research indicates resveratrol suppresses the effect of triggers such as collagen, thrombin, and oxidized LDL on platelet activation and aggregation.266-268 One study found resveratrol inhibited platelet aggregation and fibrinogen adhesion induced by the stress hormone epinephrine.269 Some clinical evidence shows a potential role for resveratrol in protecting cardiovascular and metabolic health and improving conditions associated with thrombosis risk.264
Ginseng
Extracts from Chinese ginseng (Panax notoginseng) are used intravenously in China to treat acute thrombosis, and some evidence suggests oral extracts from ginseng species may also have antithrombotic benefits. Biologically active saponins, known as ginsenosides, are present in Chinese ginseng as well as red ginseng (Panax ginseng) and American ginseng (Panax quinquefolium).270-272 An oral formulation of ginsenosides was found in a 28-day clinical trial to reduce platelet aggregation.273 Preclinical research suggests ginsenosides can inhibit platelet expression of adhesion molecules, platelet aggregation, and platelet reactivity to thrombotic triggers, which may confer protection against excessive blood clotting.274,275 In addition, ginsenosides have been shown in laboratory and animal studies to reduce vascular inflammation and stiffness, stabilize atherosclerotic plaques, and slow atherosclerosis progression, which may help prevent thromboembolism.276 Ginsenosides may also improve vascular function by reducing free radical production, increasing nitric oxide, and regulating lipid levels.270 Ginseng and ginsenosides have anti-inflammatory and broad health-promoting effects that may benefit individuals with conditions associated with thrombosis risk.270,277 Other compounds from ginseng may further contribute to its antiplatelet activity.278
Curcumin
Curcumin, a carotenoid found in the culinary spice turmeric, possesses oxidative stress-reducing and anti-inflammatory properties. Its potential antithrombotic effects have been demonstrated in laboratory and animal studies.279,280 Both antiplatelet and anticoagulant actions have been reported.279 Findings from a preclinical study indicate curcumin may be helpful in promoting endothelial repair and the resolution of venous thrombosis.281 In mice, treatment with curcumin prevented the rise in inflammatory factors, blood pressure, and D-dimer seen with exposure to air pollution (diesel exhaust), a common environmental factor known to increase inflammation and blood clot risk.282
Guavirova
The South American plant guavirova (Campomanesia xanthocarpa) has been used traditionally to treat high cholesterol levels, obesity, and various inflammatory conditions.283 In preclinical research, guavirova extract reduced platelet aggregation, prolonged coagulation time, stimulated fibrinolysis, and decreased inflammatory signaling in endothelial cells.283,284 In a small controlled trial, 23 healthy adults were given either 1,000 mg of guavirova, 100 mg of aspirin, or 500 mg of guavirova plus 50 mg of aspirin daily for five days. Guavirova inhibited platelet aggregation more strongly than the blood thinner drug aspirin and had a synergistic effect when combined with aspirin.285 In two clinical trials, guavirova was found to reduce total and LDL-cholesterol levels, lower oxidative stress, and increase endothelial nitric oxide production in individuals with high cholesterol levels.286,287
7 Causes, Risk Factors & Associated Conditions
There are three overarching mechanisms, known collectively as Virchow’s triad, associated with thrombosis risk: 1) disturbed blood flow, including stasis (stoppage or slowing of circulation) and turbulence; 2) endothelial (inner blood vessel) inflammation/injury; and 3) a high propensity to form clots, also known as excessive blood clotting or hypercoagulability, due to widespread platelet and clotting protein activation.3,11 All of the conditions associated with thrombosis involve one or more of these mechanisms.
Although venous and arterial thrombosis are distinct conditions, the factors and conditions that increase their likelihood overlap. Furthermore, thromboembolic conditions of the arteries and veins frequently co-occur.5,288,289 The role of platelets, through their inflammation- and coagulation-promoting effects, is increasingly understood to be central to both pathologies.290
Aging
Older age is accompanied by an increasing level of oxidative stress, which is linked to endothelial inflammation and platelet activation. In addition, levels of fibrinogen (used to make fibrin, a component of blood clots) rise with age. Fibrinogen is known to promote thrombosis. Aging is also frequently associated with blood clot symptoms and conditions that promote venous stasis, such as decreased physical activity and greater immobility.5
Chronic Venous Disease
Chronic venous insufficiency and varicose veins are characterized by weakness in the venous walls and can result in venous stasis. Stasis is a major cause of DVT, pulmonary embolism, and post-thrombotic syndrome.291 Interestingly, patients with chronic venous insufficiency may be more likely to have arterial disease, such as atherosclerosis. In one study, 17% of those with chronic venous insufficiency also had peripheral (non-coronary) artery disease, which is mainly due to atherosclerosis, and the incidence was higher in those with more severe venous disease.292
Atherosclerosis
Atherosclerosis is a primary underlying condition in arterial thrombosis and embolism.31,293 In the coronary arteries, it is also a risk factor for atrial fibrillation, a common type of arrhythmia linked to increased thromboembolism and a 5-fold increase in risk of stroke.33,294,295 In the peripheral arteries, atherosclerosis increases inflammatory signaling and changes in blood flow that contribute to thrombosis.296 Peripheral artery disease is associated with increased incidence of chronic venous insufficiency. In one study, 21% of those with peripheral artery disease showed signs of venous insufficiency on vascular imaging, and the presence of venous insufficiency was correlated with increased severity of arterial disease.297
Smoking
Smoking is an independent risk factor for arterial thrombosis, through its damaging effects on arterial health, and is a factor in hypercoagulability (excessive blood clotting). Smoking has also been associated with increased risk of venous thrombosis, pulmonary embolism, and post-thrombotic syndrome.5,288,289
Oxidative Stress
Free radicals, especially oxidized low-density lipoprotein (LDL), promote vascular inflammation, increase the expression of tissue factor on cellular surfaces, damage proteins that regulate coagulation, heighten reactivity of platelets, and disrupt antithrombotic hemostatic mechanisms, raising the risk of thrombosis.293,298,299
Abnormal Lipid Levels
Dysregulation of cholesterol, triglycerides, and dietary fatty acids results in high levels of oxidized lipids. This stimulates an increase in the number of platelets, as well as increased production of larger, prothrombotic platelets, in the blood and sensitizes platelets so that they are more easily activated. The risk of both venous and arterial thrombosis are higher in this state.4 In addition, high concentrations of LDL increase blood viscosity, which slows blood flow and inhibits anticoagulation and fibrinolytic mechanisms.300
Diabetes
Insulin resistance and high blood glucose levels increase oxidative stress, trigger vascular inflammation and endothelial dysfunction, activate platelets and pro-coagulation proteins, and inhibit fibrinolysis. Through these mechanisms, diabetes is a major risk factor for atherosclerosis and its thromboembolic complications, including heart attack and stroke.301 In addition, diabetes has been associated with increased risk of VTE in multiple observational studies.289
Obesity
Obesity is consistently and closely linked to higher risk of atherosclerosis and its thromboembolic complications.31 People with obesity also have more than twice the risk of VTE as those without obesity. This may be related to stasis resulting from increased venous pressure or pro-inflammatory and pro-thrombotic conditions resulting from metabolic disturbance.289
High Blood Pressure
High blood pressure causes vascular injury that triggers inflammatory signaling and activates platelets.12,31 Hypertension is a known risk factor for cardiovascular events related to arterial thrombosis and has been associated with a higher likelihood of VTE.289
High Homocysteine Levels
Elevated concentrations of homocysteine appear to raise platelet activity and lipid oxidation, and have been linked to venous thrombosis as well as arterial thrombosis.181,302 High homocysteine levels are associated with increased risk of cardiovascular events related to arterial thrombosis, particularly stroke.186,302 Homocysteine has also been implicated as a contributor to atrial fibrillation, a type of arrhythmia that disrupts normal blood flow and increases the risk of clot formation.186,303
Sleep Apnea
Sleep apnea is marked by intermittent bouts of hypoxia (low oxygenation), heightened activation of the sympathetic (fight-or-flight) nervous system, and disruption of the body’s circadian control system. These conditions trigger systemic inflammation, vascular injury, and increased production of proteins involved in the clotting process, including von Willebrand factor, tissue factor, and fibrinogen.304,305 Hypoxia also interferes with the breakdown of blood clots through fibrinolysis.304,305 The procoagulatory state induced by sleep apnea may be a contributing factor in its association with an increased risk of heart attack and stroke.304
Cancer
Venous and arterial thromboembolism are major complications of cancer, especially solid tumors.306 In fact, tumor-induced thromboembolism is the second most frequent cause of death in cancer patients.307 Microparticles secreted by cancer cells have been noted to express high amounts of tissue factor, a protein that helps initiate a procoagulatory state and facilitates cancer progression and metastasis.1,307 In addition, the tumor microenvironment is rich in platelets and clotting proteins that enhance coagulation.307
Hospitalization and Surgery
Hospitalization and surgery are often associated with physical trauma, critical illness, and prolonged immobility, and greatly increase the risk of venous stasis, DVT, pulmonary embolism, and post-thrombotic syndrome.16,19 Venous thrombosis can also be related to use of venous catheters, intravenous medications, and venous blood draws.9 Pulmonary embolism is the most common cause of preventable death in hospitalized patients.18 In addition, stasis, as well as increased propensity to form clots due to trauma or infection, result in greater risk of arterial thrombosis and stroke. As many as 17% of all strokes affect patients hospitalized for other diagnoses or procedures.308 Intravenous administration of clot busters may be used in severe cases of thrombosis in hospitalized patients.
Prolonged Inactivity
Long periods without physical activity, such as long car rides or air travel, increase susceptibility to venous thrombosis and pulmonary embolism in people with other risk factors.309,310 Over time, sedentary behavior also contributes to metabolic disturbances that increase risk of arterial thrombosis.293
Prolonged computer use in particular has been recognized as a new major cause of inactivity and a risk factor for thrombosis, giving rise to a condition termed “e-thrombosis,” or computer-related thrombosis.311 While most risk factors for thrombosis affect mainly the elderly, computer-related thrombosis tends to occur in younger individuals. Cases have even been reported in adolescents who play video games for extended periods of time.312 Avoiding long, uninterrupted, seated immobility while using a computer as well as wearing non-restrictive clothing and using comfortable sitting positions and ergonomically-designed work stations may help mitigate computer-related thrombosis risk.311
Thyroid Disease
High levels of thyroid hormone, which occur in hyperthyroidism, activate pro-coagulation pathways and reduce fibrinolytic activity. Hyperthyroidism increases risk of DVT, post-thrombotic syndrome, and pulmonary embolism, as well as the likelihood of arterial thromboembolism, in part by triggering atrial fibrillation and flutter.313,314 Hypothyroidism has the opposite effect on hemostasis and raises risk of bleeding and hemorrhage; however, it can also be a risk factor for thromboembolism.314-316
Infection, Transplant, and Transfusion Reactions
In some cases, acute immune reactions can escape control mechanisms leading to a hyperinflammatory syndrome and overactivation of coagulation pathways that may cause excessive blood clotting. This hypercoagulable state is sometimes called thrombophilia and is associated with severe illness and poor outcomes. Acute viral infections such as influenza increase the risk of thromboembolic coronary events, such as heart attack, six-fold.28 Thrombophilia can also be triggered by transplant or transfusion reactions.3,11
Other Causes of Thrombophilia
Thrombophilia can also be due to inherited and non-inherited conditions that affect hemostasis. Among the inherited conditions are rare genetic deficiencies in anticoagulant proteins.3 Autoimmune diseases like inflammatory bowel disease and anti-phospholipid syndrome, as well as liver disease, kidney disease, pregnancy, and the use of oral estrogens (eg, hormone replacement therapy or high-dose oral birth control pills) are among the many potential causes of non-inherited thrombophilia.3 However, low-dose estrogens (<50 mcg/day), such as those typically used in transdermal postmenopausal hormone replacement therapy or low-dose oral birth control pills, do not appear to increase risk of thrombosis. The transdermal route of administration in general appears to be a safer option with regard to thrombosis risk.317,318 Heparin-induced thrombocytopenia is an unusual condition that occurs in some people being treated with the anticoagulant heparin. In heparin-induced thrombocytopenia, an immune reaction to heparin causes breakdown of platelets, releasing their contents and triggering widespread platelet activation, initiation of coagulation pathways, and a dramatic increase in risk of thrombosis.319
8 Blood Clot Signs & Symptoms
Blood clot symptoms associated with thromboembolism are related to blood flow obstruction and depend on the location of the blood clot.
Superficial venous blood clots typically cause redness and tenderness along a cord of skin over the affected vein segment.9 At least 50% of patients with DVT have no blood clot symptoms, but those who do frequently experience swelling, cramping pain, warmth, and redness near the blockage, usually in a calf or thigh.3,327 They may also describe difficulty moving the limb and pain that radiates away from the site of the blood clot.3
A pulmonary embolism is generally marked by symptoms such as sharp chest pain with breathing, shortness of breath, fatigue, back pain, and fainting. Rapid breathing, rapid heart rate, fever, and low oxygen saturation may be noted on physical exam. In severe cases, pulmonary embolism can quickly evolve into a life-threatening emergency.3,6
Arterial thrombosis becomes symptomatic when a coronary, cerebral, or another critical artery becomes occluded. The classic blood clot symptoms of angina or heart attack caused by acute thrombosis of a coronary artery include crushing left-sided chest heaviness or pain, radiating to the left arm or jaw. Pain described as stabbing or burning in the epigastric region or back also frequently occur, especially in women.3,328 Acute thromboembolism affecting a cerebral artery is a cause of transient ischemic attack or stroke, which are associated with a range of symptoms such as confusion, headache, vision changes, weakness, difficulty walking or moving the extremities, difficulty swallowing, and unusual nerve sensations.3 Thrombosis and embolism can affect other arteries, including lower limb, mesenteric, renal, and retinal arteries, resulting in site-specific symptoms.27,329
9 Diagnosis of Blood Clot-Related Conditions
Venous Thrombosis
Screening. The first step in diagnosing DVT is to assess its likelihood using history and physical exam findings. A scoring system called the Wells score is an accepted tool for assessing DVT probability. The Wells score is determined by major risk factors (recent history of surgery or bed rest, immobilization, or presence of cancer), physical exam findings (edema, engorgement, enlargement, tenderness, and superficial vein dilation in the affected limb), and history (previous DVT and absence of another likely explanation for signs and symptoms).330 If the Wells score indicates DVT is likely, further assessment may be warranted.
Another screening test used in assessing thrombosis is D-dimer. D-dimer is a byproduct of fibrinolysis, and levels are elevated in patients with thrombosis due to the simultaneous activation of thrombotic and fibrinolytic pathways.330 A normal D-dimer level indicates a diagnosis other than thrombosis; however, an elevated D-dimer level is not necessarily diagnostic, since higher levels are seen in patients with infection or cancer, those who have recently undergone surgery or experienced a physical trauma, during pregnancy, and with aging.3,330 A high D-dimer level, along with a high Wells score, supports further diagnostic work-up. A high level of C-reactive protein, a marker of systemic inflammation, increases the likelihood of a thrombosis diagnosis.331
Imaging. Compression ultrasound (CUS), which is used as first-line imaging in most patients with suspected DVT,330 is performed by applying moderate probe pressure during ultrasound along a vein to assess compressibility. A lack of compressibility indicates the presence of a blood clot.6 CUS is highly accurate for diagnosing the more dangerous DVTs affecting proximal veins, but is less accurate in identifying distal DVTs. In some cases, a repeat CUS five to seven days after a negative result is recommended to detect distal DVTs that may have become proximal, and therefore pose an increased risk for pulmonary embolism.330 Computed tomography venography (CTV) and magnetic resonance venography (MRV) appear to have similar accuracy to CUS for diagnosing DVT, but are not widely used because of their invasive nature (ie, since they involve intravenous injections of contrast solutions).6,330 CTV and MRV are generally reserved for cases when CUS is less reliable or not possible, such as those with severe obesity or in a cast, or individuals with suspected thrombosis in a pelvic or abdominal vein.330
Pulmonary Embolism
A set of clinical criteria is used to assess the likelihood of pulmonary embolism. D-dimer level may be checked in clinically uncertain cases. If pulmonary embolism is suspected, the diagnosis can be confirmed using computed tomography pulmonary angiography (CTPA).6
Arterial Thrombosis
Screening. Standard tests for cardiovascular risk can be considered indicators of arterial thrombosis risk. These include total, HDL-, and LDL-cholesterol, triglyceride, glucose, and homocysteine levels, as well as hemoglobin A1c, blood pressure, and waist circumference.12 An elevated level of high-sensitivity C-reactive protein also suggests an increased risk of thromboembolic events like heart attack and stroke, and may be used to monitor progression of atherosclerosis.332,333 In addition, fibrinogen level is a more specific indicator of thrombosis risk, since excess fibrinogen in circulation increases the likelihood of clot formation.334
Although D-dimer levels are not routinely measured to diagnose arterial thrombosis, some evidence suggests they may be useful as an indicator of arterial thromboembolic risk. A study that followed 7,863 subjects with a history of heart attack or unstable angina for six years found higher D-dimer levels were associated with a higher risk of major coronary artery and cardiovascular events, as well as VTE. Ten years later, elevated D-dimer levels were found to be linked to increased cardiovascular, cancer, and all-cause mortality, independently of other risk factors.335 D-dimer levels are increasingly being used to help with treatment decisions and assess risk of recurrence in stroke patients.336
Imaging. Multidetector computed tomography (MDCT) angiography is a first-line non-invasive procedure used to assess risk of thrombosis by visualizing arterial plaque and its calcium content. This technology allows for rapid and high-resolution imaging of the walls of the carotid and coronary arteries, showing narrowing of the inner diameter (lumen) and providing an estimated plaque volume. Furthermore, plaque characteristics that suggest vulnerability to thrombosis, such as evidence of vascular remodeling, spotty calcification, and lower plaque density, can be detected with MDCT angiography.7
Carotid ultrasonography is a first-line method that uses sound waves to detect narrowing and distortion of the carotid artery due to plaque, as well as qualities of plaque, such as greater thickness, low plaque density, spotty calcification, and ulceration, that indicate greater vulnerability following a stroke or TIA. Doppler ultrasound can be used to observe blood flow, such as in the cerebral vessels, where it can detect particles traveling in the cerebral bloodstream that may cause ischemia.7,338 Ultrasound evaluation of atherosclerotic plaques can be further enhanced by the use of injected contrast agents.338
Magnetic resonance imaging (MRI) is sometimes used to visualize the carotid arteries. The advantage of MRI lies in its ability to detect features of the fibrous cap of a plaque that indicate its risk of rupture. An MRI can also show bleeding at the site of a ruptured plaque.7,339
Positron emission tomography (PET) is an invasive nuclear imaging test that involves the use of an injected tracer to evaluate arteries affected by atherosclerosis. PET for this purpose is still in its investigational stage.7,339
Intravascular ultrasound (IVUS) is performed by inserting an ultrasonic device into the arterial system using a catheter. Sound waves emitted by the device can show some characteristics of the inside of the blood vessel walls, including thickness, indicative of plaque.7,338,339
Optical coherence tomography uses reflected near-infrared light to produce high-resolution images of the inner walls of arteries. It can detect the presence of plaque, features linked to risk of plaque rupture, and evidence of rupture and thrombosis. Near-infrared spectroscopy (NIRS) measures the absorption of near-infrared light to locate the exact position and density of lipids in the vessel wall. It has been suggested these modalities may provide the most useful information when used together or with other imaging technologies.7,339
Clotting Disorders
Tests for underlying causes of thrombophilia may be performed in individuals who experience a blood clotting event without relevant risk factors. Thrombin time (TT) is a non-specific test of clotting capacity that measures how long it takes for a blood clot to form in a plasma sample to which thrombin has been added. If thrombin time is low, tests for autoimmune diseases, liver disease, cancer, and other conditions associated with increased clotting risk may be indicated. Protein C, protein S, and antithrombin levels can be measured to screen for clotting disorders caused by inherited deficiencies of these coagulation inhibitors.2
10 Treatment of Blood Clots & Thrombosis
In a thromboembolic emergency, thrombolytic agents that promote fibrinolysis are often used, sometimes in conjunction with a medical ultrasound technique called sonothrombolysis. Surgical removal of the blood clot, known as thrombectomy, is sometimes performed to remove thrombi from accessible large arteries or veins, but is not possible for other locations.8 Because of the devastating nature of thromboembolic events, prevention using long-term medical therapy with antiplatelet or anticoagulant drugs (blood thinners) is the standard approach in those at highest risk. It is important to note that none of these medications address the underlying conditions that cause thrombosis and all alter hemostatic control of blood clotting, increasing the risk of bleeding with potentially dangerous outcomes.3 In addition to drugs, compression stockings are sometimes recommended to those who have experienced DVT.340
Thrombolytic Therapy
Thrombolytics, also known as plasminogen activators or “clot busters,” are used in the acute stage of a thromboembolic event to quickly dissolve dangerous intravascular clots and restore blood flow. These drugs are administered intravenously within several hours of the onset of acute heart attack, stroke, DVT, pulmonary embolism, peripheral artery occlusion, occlusion of an implanted catheter, or a blood clot in the heart.341 Thrombolytics promote the production of the fibrinolytic protein, plasmin, from plasminogen in the fibrin matrix of a clot. FDA-approved clot busters include alteplase (Activase), urokinase (Kinlytic), and streptokinase (Streptase). A number of other plasminogen activators are currently under investigation.8,341
In addition to the substantial risk of unintended bleeding, these drugs sometimes cause low blood pressure, allergic reactions, and arrhythmias. The risk of bleeding is especially high in those who are elderly, had a recent stroke or surgery, have uncontrolled hypertension, or have a bleeding tendency, and is compounded in those who regularly use anticoagulant medications.341
Sonothrombolysis
Sonothrombolysis uses ultrasound to generate clot-breaking sound waves. It is mainly used in conjunction with thrombolytic drugs and has been found to make thrombolytic treatment faster and more effective in some circumstances.342 Sonothrombolysis is thought to work by creating acoustic forces that can displace and loosen clots, facilitating thrombolytic drug diffusion into clots. This may potentially allow for lower thrombolytic drug exposure and reduced risk of hemorrhage.343
Antiplatelet Therapy
Antiplatelet drugs, a class of blood thinner medications that inhibit platelet activation and aggregation, are typically used in long-term prevention of arterial thrombosis.
Aspirin is a common anti-inflammatory antiplatelet medication that works by inhibiting cyclooxygenase, an enzyme that participates in inflammatory and pro-coagulation pathways. Low-dose aspirin (75‒100 mg per day) is widely recommended as a long-term preventive strategy in patients with a history of stroke or heart attack, in whom it has been found to lower risk of a second event by approximately 18–19%.344,345 It is frequently used in combination with other antiplatelet agents that inhibit other platelet activation pathways and work additively or synergistically with aspirin.4 However, long-term use of aspirin, even at low doses, is associated with an increased risk of bleeding, most notably in the digestive tract and the brain. Since large clinical trials have found daily low-dose aspirin provides little to no cardiovascular benefit in those without a prior history of a coronary event or stroke, guidelines do not recommend its use for such individuals, except in those with exceptionally high cardiovascular risk and no other factors that increase bleeding risk.344,346
Clopidogrel (Plavix) works by inhibiting receptors on platelet membranes called purinergic P2Y12 receptors. When these receptors interact with a circulating platelet-activating molecule called adenosine diphosphate (ADP), they upregulate expression of a protein complex involved in platelet aggregation.347 Clopidogrel has been found to reduce the risk of recurrent stroke in patients who have experienced a non-disabling stroke that is not due to an embolism from a coronary artery.344,347 It is frequently used in combination with aspirin in the first 90 days after a stroke to lower the risk of recurrent events, but has not been found to add to aspirin’s protective effect in long-term prevention of recurrent stroke and adds to the risk of bleeding. Clopidogrel is considered an acceptable alternative to aspirin in patients with a history of stroke, providing a similar degree of protection against recurrent stroke.347 Side effects of clopidogrel include increased bleeding, reduced number of neutrophils (a type of immune cell), and a rare dangerous condition called thrombotic thrombocytopenic purpura in which blood clots form in small blood vessels throughout the body.4
Two other drugs in the P2Y12 receptor inhibitor class, ticagrelor (Brilinta) and prasugrel (Effient), are under investigation for their possible benefits during coronary artery stent implantation after a heart attack.4,344
Dipyrimidole (Persantine) works in part by inhibiting platelet phosphodiesterase, an enzyme that increases platelet reactivity. The combination of dipyrimidole plus aspirin may be more effective than either drug alone for preventing stroke in patients with a recent non-embolic TIA or stroke.344,347 In addition to increasing the risk of bleeding, this combination causes headaches in an estimated 40% of users.347
Cilostazol (Pletal) inhibits phosphodiesterase but has effects in both platelets and endothelial cells that line the blood vessel walls. Cilostazol inhibits platelet aggregation and promotes dilation of arteries, and is mainly used to treat peripheral artery disease.4,344
Anticoagulant Therapy
Anticoagulant drugs, also referred to as blood thinners, inhibit various parts of the clotting cascade and are used long term to prevent thrombosis in high-risk patients.
Heparin is a naturally occurring anticoagulant produced by certain types of immune cells that interacts with antithrombin, increasing its inhibiting effect on several clotting factors and thrombin.348 Heparin is available in various forms, including unfractionated heparin, low-molecular weight heparin, and synthetic analogs, all of which require intravenous or subcutaneous injection. Because of its rapid ability to block the clotting cascade, heparin is usually used for 5–10 days in the acute stage of venous thrombosis to reduce expansion of the thrombus and prevent pulmonary embolism.349,350 It may also be prescribed for long-term use in cancer-related thrombosis.349 However, heparin is often unable to completely resolve blood clots. Low platelet numbers can occur with heparin use, increasing the risk of bleeding.350
Warfarin (Coumadin) is a vitamin K antagonist and until recently was the only oral anticoagulant in use. It works by inhibiting vitamin K recycling, thereby reducing activation of vitamin K-dependent coagulation factors.350 Warfarin has a long half-life in the body, but in circumstances requiring rapid reversal of the anticoagulant effect, such as hemorrhage, overdose, or the need for emergency surgery, warfarin’s effects can be overridden by one of three antidotes: vitamin K, prothrombinase complex concentrate, or fresh frozen plasma.351
Effective anticoagulant therapy using warfarin is especially challenging, in part because the therapeutic window—the dose range at which the likelihood of benefit from blood clot reduction outweighs the likelihood of harm from bleeding—is narrow. Responsiveness to warfarin therapy varies due to genetic differences that impact its metabolism. Because the effective dose depends partly on the blood concentration of vitamin K, dietary changes also affect responsiveness. Furthermore, warfarin is prone to interactions with foods unrelated to their vitamin K content, herbs, and other drugs. For these reasons, long-term warfarin use requires frequent monitoring, often leading to dose adjustments, to maintain both effectiveness and safety.350
Direct oral anticoagulants (DOACs) are a class of anticoagulant medications that either inhibit thrombin or factor Xa.
- Inhibiting thrombin decreases the conversion of fibrinogen to fibrin, an essential component of clot formation.
- Inhibiting factor Xa decreases the conversion of prothrombin to thrombin.
As of mid-2024, there are five DOACs approved for use in the United States:
- Betrixaban (Bevyxxa)
- Rivaroxaban (Xarelto)
- Apixaban (Eliquis)
- Edoxaban (Savaysa)
- Dabigatran (Pradaxa)
The mechanism(s) by which DOACs work differ from those of two other common anticoagulants:
- Warfarin inhibits clotting by acting as a vitamin K antagonist, and
- Heparin/low-molecular weight heparin works by activating antithrombin, which inactivates the coagulation enzymes thrombin, factor Xa, and factor IXa.
Widespread use of DOACs began in 2010 due to multiple factors, including421:
- Standardized dosing
- Decreased frequency of follow-ups
- Less adverse effects
- Fewer medication and food interactions
- Quicker onset of action
Because of these factors, DOACs are generally preferred over warfarin by both patients and physicians. DOACs are FDA approved for the following conditions359:
- Stroke prevention in non-valvular atrial fibrillation (NVAF)
- Prevention of thromboembolism after total hip and knee replacement
- Treatment of DVT and pulmonary embolism
- Prevention of recurrent DVT and pulmonary embolism
- Prevention of thromboembolisms in hospitalized, acutely ill patients
- Prevention of major cardiovascular events in patients with chronic coronary artery disease and peripheral artery disease
Despite their many advantages over warfarin, there are contraindications for the use of DOACs, including the following:
- Having a mechanical (versus a bioprosthetic) heart valve
- Moderate-to-severe mitral stenosis
- Severe kidney and liver disease
- Gastrointestinal absorption issues from either disease states or bariatric surgery
In these instances, if anticoagulation is needed, warfarin or heparin may be recommended as a first-line anticoagulation agent.359 In addition, DOACs are more expensive—sometimes much more—than warfarin, which can be a significant barrier to access if patients do not have insurance coverage. Despite these issues, the use of DOACs has grown since their inception, now accounting for approximately 50% of all oral anticoagulants used in the United States.422
Comparative Safety & Efficacy: DOAC vs. DOAC
All five DOACs have similar safety profiles. A 2018 review concluded that apixaban was the safest, followed by dabigatran and rivaroxaban.423 A cohort study published in 2023 examined the long-term comparative effectiveness and safety of dabigatran, rivaroxaban, apixaban, and edoxaban in patients with atrial fibrillation.424 In this study, all the DOACs examined showed similar stroke and systemic embolism risks. In terms of major bleeding or clinically relevant non-major bleeding risks, dabigatran and apixaban had significantly lower risks when compared to rivaroxaban and edoxaban. Higher mortality was seen in diabetic patients using apixaban when compared to dabigatran and edoxaban.
Drug-drug interactions directly influence the safety profile of medications. For DOACs, dabigatran, edoxaban, and betrixaban have the fewest drug-drug interactions, followed by apixaban and rivaroxaban.424 In the context of kidney disease, a standard dosage of apixaban can be used, while dabigatran, rivaroxaban, and edoxaban should be used at lower doses.425
The use of any anticoagulant, including DOACs, can lead to an increased risk of gastrointestinal bleeding. Once- or twice-daily dosing does not appear to affect bleeding risk; a 2021 meta-analysis examining the differences between once- and twice-daily dosing of DOACs reported that there was no significant difference between the different drugs in terms of major thrombotic events or major bleeding risks.426
In general, apixaban appears to have the lowest risk of bleeding. A 2025 cohort study that followed over 160,000 American patients with VTE found that apixaban was associated with a ~30% lower risk of major bleeding compared with warfarin and rivaroxaban. Apixaban also reduced the risk of recurrent VTE compared with warfarin and rivaroxaban.450 A Swedish nationwide registry-based study published in 2023 examined the association of types of anticoagulants and risk of bleeding among 45,114 patients with VTE during the first six months of treatment, and then from six months to five years.427 The authors concluded that apixaban had a lower risk in terms of bleeding than rivaroxaban during the first six months of treatment. From six months to five years, the bleeding risk was similar. In a different network meta-analysis that included over 40,000 patients with atrial fibrillation and valvular heart disease, apixaban had a lower risk of major bleeding events compared with the other DOACs and warfarin.428
A systematic review and meta-analysis of 19 randomized controlled trials examined the risks of non-major bleeding events between different DOACs. The study showed that apixaban had the lowest risk, followed by dabigatran, rivaroxaban, and edoxaban.429
While there have not been head-to-head randomized controlled trials comparing the efficacy of various DOACs to each other, several observational studies and meta-analyses have been conducted. In a systematic review and meta-analysis on the safety and efficacy of DOACs and warfarin in elderly patients with atrial fibrillation, researchers concluded that there were no significant differences in the DOACs studied (rivaroxaban, dabigatran, apixaban, and edoxaban) in terms of stroke or blood clot prevention.430 In an observational study of 321,501 patients with nonvalvular atrial fibrillation, apixaban, rivaroxaban, and dabigatran comparably decreased the risk of stroke and systemic thromboembolic events.431 A retrospective study that included nearly 142,000 patients with atrial fibrillation found that risk of mortality and stroke was lower in patients treated with rivaroxaban compared to apixaban. Rivaroxaban also had lower rates of intracranial bleeding compared to apixaban, but higher rates of gastrointestinal bleeding. There was no significant difference in rates of heart attacks or overall bleeding between the different DOACs, which included rivaroxaban, apixaban, and dabigatran.432
Approximately 1.5 million people a year in the United States have some type of joint replacement surgery. Studies show that 0.6–3.0% of those patients will suffer a DVT and/or pulmonary embolism. A 2023 retrospective cohort study examined the risk factors for post-surgical DVT and pulmonary embolism occurrence among 29,264 patients who received DOACs (apixaban or rivaroxaban) or aspirin. The results showed that underlying patient risk factors, but not the choice of aspirin or DOAC, were associated with developing a DVT or pulmonary embolism.433
A randomized clinical trial of 601 Japanese men and women with cancer and DVT, and a mean age of about 71 years, examined whether a longer duration of edoxaban therapy would be useful in DVT prevention. The researchers showed that a longer treatment period—12 months vs. three months—was superior in preventing recurrent DVTs or DVT-related deaths.434
Coronary artery disease is a leading cause of death and disability. The COMPASS study examined the effects of using a DOAC for secondary prevention of major cardiovascular events in patients with either coronary artery disease or peripheral artery disease. In this randomized trial, 27,395 women and men were given either low-dose rivaroxaban plus aspirin, rivaroxaban alone, or aspirin alone. After a mean follow-up time of two years, results showed that there was a reduction in heart attacks and strokes in the group of patients that were taking rivaroxaban and aspirin compared with the patients only taking aspirin. There was no greater risk of intracranial or fatal bleeding in the rivaroxaban and aspirin group.435
Comparative Safety & Efficacy: DOACs vs. Warfarin
Atrial fibrillation is a common cardiac issue, affecting 10% of people 75 or older. Atrial fibrillation can cause blood clots, strokes, and heart failure. Most patients with atrial fibrillation are recommended anticoagulant therapy since studies show this will significantly decrease the risk of clots and strokes.429
A meta-analysis of 22 studies comprising 440,281 patients examined the safety and efficacy of DOACs compared to warfarin in elderly patients with atrial fibrillation. Patients taking DOACs versus warfarin had a significantly lower risk of strokes and blood clots as well as a reduction in the risk of fatal bleeding and intracranial bleeding, but an increased risk of gastrointestinal bleeding.430
Another study examined the safety of switching from warfarin to a DOAC in frail elderly patients with atrial fibrillation. In this open-label, randomized, controlled trial, 1,323 men and women with a mean age of 83 were either switched from warfarin to a DOAC (662 patients) or left on warfarin (661 patients). Results showed that switching patients who were stable on warfarin to a DOAC caused more bleeding complications compared to staying on warfarin, without an associated reduction in strokes or clots.437 In a cohort study on non-frail elderly (mean age 82) men and women with atrial fibrillation in Norway, researchers showed that both standard and low-dose DOACs had a similar risk profile in regard to strokes and clots compared to warfarin, with a similar or decreased risk of bleeding issues.438
Heart valve replacement surgery is becoming more common as the United States population ages; over 100,000 heart valve replacements are performed every year. While non-DOAC anticoagulants are recommended for people with mechanical heart valves, those with bioprosthetic valves may not need treatment. However, for those who do, studies have shown that DOACs may be advantageous over warfarin. A meta-analysis of 28 studies with 74,660 patients who had bioprosthetic valves and atrial fibrillation showed that DOACs were more effective in reducing the risks of bleeding, strokes, systemic embolism, and intracranial bleeding when compared to warfarin.439 A similar systematic review and meta-analysis was published concerning the efficacy and safety of DOACs versus warfarin for patients with left-sided bioprosthetic heart valves and atrial fibrillation. This analysis, comprised of 13 studies and 27,993 patients, showed that DOAC usage reduced the risk of stroke by 33% compared to warfarin. Major bleeding in patients taking DOACs was reduced by 28% versus warfarin. Patients in this study who were less than 75 years old and on DOACs had a 45% reduction in stroke rate.440
In the context of atrial fibrillation, a systematic review and meta-analysis of 21,185 patients with non-valvular atrial fibrillation with valvular heart disease compared DOACs and warfarin for prevention of clots and strokes. Researchers showed that both strokes and clots were less frequent in patients taking DOACs; there was also a lower incidence of intracranial bleeding compared with patients taking warfarin.441
Chronic kidney disease is a known risk factor for atrial fibrillation. A meta-analysis of eight randomized controlled trials and 46 observational studies was done to evaluate the efficacy and safety of DOACs in patients with atrial fibrillation and chronic kidney disease. Results of the study showed that DOACs were superior to warfarin in terms of preventing thromboembolic events, as well as reducing bleeding risks, in patients with chronic kidney disease.442
Finally, DOACs appear to be more efficacious in people with peripheral artery disease, which is a significant cause of limb loss. A 2023 systematic review and meta-analysis examined the effect of DOAC usage in individuals with peripheral artery disease. Results of studies analyzed showed that in people with peripheral artery disease and non-valvular atrial fibrillation, use of DOACs significantly reduced the risk of limb loss, strokes, and systemic embolism compared with using warfarin.443
Comparative Safety & Efficacy: DOACs vs. Low-Molecular Weight Heparin
People with cancer have a four- to seven-fold increased risk of developing DVT and pulmonary embolism. In a cohort study of 1,109 male and female Asian patients with cancer-related DVT and pulmonary embolism, those taking DOACs had a similar risk for recurrent DVT or pulmonary embolism when compared with those patients taking low-molecular weight heparin, but had a lower risk of gastrointestinal bleeding.444
The safety of DOACs versus low-molecular weight heparin for extended clot prevention after abdominal and pelvic cancer surgery was investigated in a systematic review and meta-analysis. Data from nine studies (two randomized controlled trials and seven observational studies) with 2,651 patients was analyzed. Patients taking DOACs had a similar incidence of thromboembolism, major bleeding events, and clinically relevant non-major bleeding events when compared with patients taking low-molecular weight heparin.445
Another systematic review and meta-analysis also examined the efficacy and safety of DOACs versus low-molecular weight heparin for blood clot prevention after cancer surgery. Examining data from 10 studies with 3,054 patients, authors of this study concluded, as in the previous study, that patients taking DOACs had a comparable risk of thromboembolism, major bleeding events, and clinically relevant non-major bleeding events when compared with patients taking low-molecular weight heparin.446
Chronic kidney disease is known to increase the risk of VTE. A systematic review and meta-analysis was done examining the efficacy of oral anticoagulants and low-molecular weight heparin to prevent VTE in patients with chronic kidney disease. Ten phase 3, randomized, controlled trials were utilized for the meta-analysis, with data from 10,840 patients evaluated. The authors of the study showed that there was no difference in efficacy between low-molecular weight heparin and DOACs in terms of preventing VTE for patients with chronic kidney disease at any level of renal impairment.447
A 2023 study examined whether DOACs are noninferior to low-molecular weight heparin in terms of preventing recurrent VTE events in people with cancer. In a randomized clinical trial that included 638 individuals with cancer and a new VTE, researchers showed the patients taking DOACs had a recurrent VTE rate of 6.1% at six months versus 8.8% for patients taking low-molecular weight heparin.448
Researchers analyzed data from the Observational Study in Cancer-Associated Thrombosis for Rivaroxaban-United States cohort (OSCAR-US) comparing the effectiveness of rivaroxaban to low-molecular weight heparin in regard to VTE risk. At three months and six months, patients taking rivaroxaban had a reduced risk of VTE compared with patients taking low-molecular weight heparin. However, at 12 months, there was no difference between those taking rivaroxaban and low-molecular weight heparin.449
Compression Stockings
Graduated compression stockings, which apply the greatest amount of pressure at the ankle and the least at the knee, are sometimes used to prevent post-thrombotic syndrome in DVT patients and relieve symptoms in patients with chronic venous disease.373 One systematic review of data from eight trials found the use of compression stockings during air travel reduced edema and VTE, which may lead to post-thrombotic syndrome, in high-risk individuals.374 However, clinical trials assessing their general efficacy have so far yielded mixed results, and many patients stop using compression stockings due to discomfort.340,375
11 Monitoring Blood Clot Therapy
Several tests are available for assessing the risk of recurrent thrombosis and monitoring long-term anticoagulant therapy.
- Activated partial thromboplastin time (APTT) assesses the function of a group of clotting proteins and fibrinogen. It can be used to monitor the effects of anticoagulation therapy, mainly in those receiving unfractionated heparin.2,348 A low APTT value in the absence of anticoagulant therapy indicates an increased propensity for clotting. Some anticoagulant medications, including unfractionated heparin, increase APTT, reflecting prolongation of the time needed for clot formation.2
- Prothrombin time (PT) evaluates the function of the clotting cascade and international normalized ratio (INR) expresses it in standardized terms. PT measures the time to form a clot under certain laboratory conditions, while INR is a calculation based on the individual’s PT value and average PT values in order to standardize for differences in laboratory methods.2 In those being treated with warfarin, PT and INR testing is performed regularly to make dosage adjustment decisions.348
- Diluted thrombin time and ecarin clotting time are used to directly assess the anticoagulation effects of dabigatran. Routine monitoring is not needed, but these tests may be helpful in circumstances such as before emergency surgery, when drug-drug interactions are in question, or in the event of a hemorrhage.376
- Anti-Xa assay measures inhibition of the clotting cascade protein Factor Xa and is used to assess the treatment effects of Factor Xa inhibitors like apixaban, rivaroxaban, and edoxaban. However, the clinical usefulness of anti-Xa assays in patients using these drugs has not been established.359,376 It may provide helpful information in those receiving heparin.348
There is no generally accepted method for routine monitoring of antiplatelet therapy. However, a variety of platelet function tests may be useful under certain circumstances, such as optimizing timing of urgent surgery after oral antiplatelet drugs are stopped or individualizing care for an actively bleeding patient. Some examples of these tests include377-379:
- Lab-based tests include light transmission aggregometry (LTA), which can assess the effects of aspirin or clopidogrel, and vasodilator-stimulated phosphoprotein assay, which is used to assess effects of clopidogrel.
- VerifyNow P2Y12 assay and Multiplate analyzer are in-office tests used specifically for assessing clopidogrel responsiveness.
- Platelet function analyzer-100 (PFA-100) and Plateletworks assay are additional tests that can be used to assess aspirin or clopidogrel effectiveness.
12 Novel & Emerging Strategies to Prevent & Treat Thrombosis
Sonothrombolysis Innovations
New technologies are being investigated for their potential to increase the effectiveness of sonothrombolysis. The use of microbubbles and nanodroplets that amplify and focus the ultrasonic mechanical action have demonstrated promising effects in laboratory and clinical studies. An ultrasonic device mounted on a catheter can deliver ultrasound from within the blood vessel and may be helpful in treating large blood clots.343,380 High-intensity focused ultrasound and histotripsy are modified therapeutic ultrasound techniques that fractionate clots and may potentially eliminate the need for thrombolytic drugs.343
Canakinumab
Inflammasomes are large protein complexes in cells that initiate the inflammatory response by increasing the production of cytokines. In atherosclerotic blood vessels, cholesterol crystals stimulate an immune cell inflammasome called NLRP3 that then triggers a cascade of inflammatory and thrombotic signaling.381
Canakinumab (Ilaris) is a monoclonal antibody that inhibits interleukin-1β, an inflammatory cytokine produced in response to NLRP3 inflammasome activation and closely linked to cardiovascular disease.381,382 A large, randomized, placebo-controlled trial found canakinumab had beneficial effects in high-risk patients. The trial included 10,061 subjects who had experienced a heart attack 30 days or more prior to enrollment and had hs-CRP levels of 2.0 mg/L or higher, indicating ongoing inflammation despite standard medical therapy for their high cardiovascular risk. They received 50, 150, or 300 mg canakinumab, or placebo as subcutaneous injections every three months for a median of 3.7 years. Those receiving 150 mg canakinumab had a 15% lower risk of heart attack, stroke, or death from cardiovascular causes compared with placebo.383 A secondary analysis of the results showed, among subjects receiving canakinumab, those who achieved an hs-CRP level of less than 2.0 mg/L had 31% lower rates of cardiovascular and all-cause deaths, while those whose hs-CRP levels remained 2.0 mg/L or higher had no reduction in these outcomes.384 However, canakinumab was also associated with a small increase in fatal infections such as sepsis (an infection in the blood), cellulitis, and pneumonia.385
Abelacimab
Abelacimab (previously MAA868) is a novel monoclonal antibody that binds to clotting factor XI and thereby interrupts activation of other clotting factors and production of thrombin and fibrin. In a single randomized open-label controlled trial supported by the manufacturer, 412 subjects undergoing total knee replacement surgery received a single intravenous injection of one of three abelacimab doses immediately following surgery, or subcutaneous injections of the anticoagulant enoxaparin (Lovenox) once daily after surgery. Compared with those who received enoxaparin, the risk of venous thromboembolism was lower in those who received the middle and high doses of abelacimab and no different in those who received the low dose.386 A trial comparing safety and tolerability of abelacimab to rivaroxaban in atrial fibrillation patients is currently underway.387
Repurposing Statins
Statins are a family of medications used to lower high cholesterol levels. Examples of statins are atorvastatin (Lipitor), lovastatin (Mevacor), simvastatin (Zocor), and rosuvastatin (Crestor). Multiple randomized controlled trials have shown their use lowers the risk of arterial thromboembolic events, including heart attack and stroke, and observational studies have reported correlations between statin use and lower DVT/pulmonary embolism risk.388-391 A meta-analysis that included data from 12 observational studies found venous thromboembolism (VTE) patients being treated with a statin drug for high cholesterol levels were 24% less likely to experience a recurrence of either DVT or pulmonary embolism.392 In one study that followed 980 elderly participants with a previous DVT or pulmonary embolism for a median of 2.5 years, statin use was associated with a 50% reduction in risk of a recurrence, but only during periods when anticoagulant drugs were not being used.393 Some evidence suggests statin users are less likely to develop post-thrombotic syndrome, a complication of DVT.394
Several mechanisms may contribute to statin drugs’ antithrombotic effects. A meta-analysis of randomized controlled trials found statins reduced levels of plasminogen activator inhibitor-1, resulting in increased production of plasmin, a fibrinolytic enzyme.395 Other research has shown statins can reduce the expression of an adhesion molecule involved in platelet aggregation, inhibit steps in the clotting cascade, and increase production of nitric oxide synthase, an enzyme involved in regulating vascular function.394,396 Statins may also decrease inflammatory marker levels, oxidative stress, platelet reactivity, and endothelial injury.396,397 The most common adverse side effects of statin therapy include muscle pain and increased blood levels of liver enzymes, and some drugs in the statin family are linked to increased risk of type 2 diabetes; uncommonly, statins can cause serious muscle tissue injury.398 Because statin use lowers CoQ10 production, patients taking statins may benefit from supplementing with CoQ10.136,138,140
Repurposing Colchicine
Colchicine (Mitigare, Colcrys) is an anti-inflammatory drug used to treat gout. In observational studies, gout patients treated with colchicine were noted to have fewer heart attacks, strokes, and TIAs, and had lower mortality than gout patients who were not treated with colchicine.399
In a randomized placebo-controlled trial, 5,522 patients with atherosclerosis receiving standard cardiovascular risk-lowering therapies were given 0.5 mg of colchicine or placebo daily. After an average of 28.6 months, the incidence of major cardiovascular events was 31% lower in those given colchicine.400 Another randomized placebo-controlled trial in 4,745 patients with a recent history of heart attack found 0.5 mg colchicine daily lowered the risk of major cardiovascular events by 23% during a median follow-up of 22.6 months; however, pneumonia occurred more frequently in those receiving colchicine.401 A meta-analysis of five randomized controlled trials with a total of more than 11,000 participants found colchicine reduced the risk of major thromboembolic cardiovascular events in patients with coronary artery disease.402 Another meta-analysis of nine trials with a total of 6,630 participants found colchicine lowered the risk of stroke, but not other major cardiovascular events.403
Colchicine inhibits the pro-inflammatory activity of the NLRP3 inflammasome protein complex, and has been found to reduce expression of tissue factor, a protein with a pivotal role in initiating thrombosis, in response to oxidized LDL-cholesterol.404-406 The most common adverse side effects caused by colchicine are nausea, vomiting, and diarrhea. In rare cases, it can cause liver injury, severe muscle injury, allergy, and blood disorders.405
Repurposing Metformin
Metformin is a blood glucose-lowering medication used to treat type 2 diabetes. It has been shown to reduce cardiovascular risk in diabetic individuals, independently of its effect on blood glucose levels. Observational evidence from a study that monitored nearly 15,000 participants with type 2 diabetes for over three years noted metformin was correlated with reduced risk of deep vein thrombosis.407 Another study comparing outcomes in more than 32,000 diabetic subjects who had undergone knee replacement surgery found those who were taking metformin had better outcomes and lower risk of complications, including DVT.408 Preclinical and clinical research has shown metformin use is associated with improved endothelial function, lower levels of tissue factor, reduced levels of oxygen free radicals, and decreased platelet activation and aggregation.409,410
Repurposing Pentoxifylline
Pentoxifylline (Pentopak, Pentoxil, or Trental) is a drug that reduces blood viscosity and improves blood flow. It is approved for use in treating claudication, a peripheral vascular disorder affecting the legs that is marked by ischemic pain, and is sometimes used to treat venous ulcers in the legs. Pentoxifylline stimulates fibrinolysis, inhibits platelet aggregation and adhesion, and reduces inflammatory cytokine and free radical production.411 Early observations suggested pentoxifylline may be helpful in treating stroke patients.412 In one trial that included 97 participants, pentoxifylline was found to be more effective than a combination of aspirin plus dipyridamole (Persantine) for preventing re-closure of blood vessels in the legs during six months following vascular surgery (for treatment of occlusion in the aortoiliac or femoropopliteal regions or both).413 A placebo-controlled trial in 51 hemodialysis patients found pentoxifylline prevented blood clots from forming in arteriovenous shunts implanted to facilitate dialysis.414 Pentoxifylline was also found to lower the risk of a dangerous blood clotting disorder in premature newborns,415 and appeared to be helpful in treating a similar clotting emergency in adult patients with severe blood infections.416 In a clinical trial comparing the effects of various antiplatelet agents, pentoxifylline was among the drugs shown to enhance the effectiveness of anticoagulant therapy in severe or recurrent DVT patients.417
Side effects of pentoxifylline include digestive upset, dizziness, headache, and flushing; less commonly, it causes chest pain, arrhythmia, or low blood pressure. Pentoxifylline can enhance the effects of blood pressure- and blood glucose-lowering medications and increase the risk of bleeding in patients taking antiplatelet or anticoagulant drugs. Because pentoxifylline increases blood flow to the heart, it is not indicated for patients with severe coronary artery disease or after acute heart attack.411
Intravenous Ginseng
Chinese ginseng (Panax notoginseng) contains saponins known as ginsenosides that are used extensively in China to treat stroke due to their ability to inhibit platelet aggregation and protect neurons from injury.275 A meta-analysis of data from 23 randomized controlled trials with a combined total of 2,196 participants found adding ginseng saponins (formulated for intravenous injection) to standard medical therapy improved outcomes in acute stroke patients.418 An intravenous preparation of Chinese ginseng saponins has also been widely studied for its ability to prevent and treat venous thrombosis, similar to Western “clot busters.” A meta-analysis of 20 randomized controlled trials with a total of 2,336 participants who had undergone orthopedic surgery due to fracture found those given injections containing ginseng saponins had fewer DVTs, lower D-dimer levels, and longer prothrombin and thrombin times.419 Another meta-analysis of 12 randomized controlled trials that included a total of 1,018 DVT patients found the use of ginseng saponin injections as an add-on therapy resulted in better treatment outcomes.420
Disclaimer and Safety Information
This information (and any accompanying material) is not intended to replace the attention or advice of a physician or other qualified health care professional. Anyone who wishes to embark on any dietary, drug, exercise, or other lifestyle change intended to prevent or treat a specific disease or condition should first consult with and seek clearance from a physician or other qualified health care professional. Pregnant women in particular should seek the advice of a physician before using any protocol listed on this website. The protocols described on this website are for adults only, unless otherwise specified. Product labels may contain important safety information and the most recent product information provided by the product manufacturers should be carefully reviewed prior to use to verify the dose, administration, and contraindications. National, state, and local laws may vary regarding the use and application of many of the therapies discussed. The reader assumes the risk of any injuries. The authors and publishers, their affiliates and assigns are not liable for any injury and/or damage to persons arising from this protocol and expressly disclaim responsibility for any adverse effects resulting from the use of the information contained herein.
The protocols raise many issues that are subject to change as new data emerge. None of our suggested protocol regimens can guarantee health benefits. Life Extension has not performed independent verification of the data contained in the referenced materials, and expressly disclaims responsibility for any error in the literature.
- O'Donnell JS, O'Sullivan JM, Preston RJS. Advances in understanding the molecular mechanisms that maintain normal haemostasis. Br J Haematol. Jul 2019;186(1):24-36. doi:10.1111/bjh.15872
- Bonar RA, Lippi G, Favaloro EJ. Overview of Hemostasis and Thrombosis and Contribution of Laboratory Testing to Diagnosis and Management of Hemostasis and Thrombosis Disorders. Methods in molecular biology (Clifton, NJ). 2017;1646:3-27. doi:10.1007/978-1-4939-7196-1_1
- Ashorobi D, Ameer MA, Fernandez R. Thrombosis. StatPearls. StatPearls Publishing Copyright © 2021, StatPearls Publishing LLC.; 2021.
- Yang M, Kholmukhamedov A. Platelet reactivity in dyslipidemia: atherothrombotic signaling and therapeutic implications. Reviews in cardiovascular medicine. Mar 30 2021;22(1):67-81. doi:10.31083/j.rcm.2021.01.256
- Poredoš P. Interrelationship between venous and arterial thrombosis. Int Angiol. Aug 2017;36(4):295-298. doi:10.23736/s0392-9590.17.03820-2
- Patel H, Sun H, Hussain AN, Vakde T. Advances in the Diagnosis of Venous Thromboembolism: A Literature Review. Diagnostics (Basel). Jun 2 2020;10(6)doi:10.3390/diagnostics10060365
- Spacek M, Zemanek D, Hutyra M, Sluka M, Taborsky M. Vulnerable atherosclerotic plaque - a review of current concepts and advanced imaging. Biomedical papers of the Medical Faculty of the University Palacky, Olomouc, Czechoslovakia . Mar 2018;162(1):10-17. doi:10.5507/bp.2018.004
- Nikitin D, Choi S, Mican J, et al. Development and Testing of Thrombolytics in Stroke. J Stroke. Jan 2021;23(1):12-36. doi:10.5853/jos.2020.03349
- Cosmi B. Management of superficial vein thrombosis. J Thromb Haemost. Jul 2015;13(7):1175-83. doi:10.1111/jth.12986
- Betts JG, Young KA, Wise JA, et al. Anatomy and Physiology 18.4 Hemostasis. OpenStax. Accessed 07/2/2021, https://openstax.org/books/anatomy-and-physiology/pages/18-5-hemostasis
- Garmo C, Bajwa T, Burns B. Physiology, Clotting Mechanism. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Available at https://www.ncbi.nlm.nih.gov/books/NBK507795/ . Last updated 09/08/2020. Accessed 05/05/2021. 2020;
- Barale C, Russo I. Influence of Cardiometabolic Risk Factors on Platelet Function. International journal of molecular sciences. Jan 17 2020;21(2)doi:10.3390/ijms21020623
- Chaudhry R, Usama SM, Babiker HM. Physiology, Coagulation Pathways. StatPearls. StatPearls Publishing Copyright © 2021, StatPearls Publishing LLC.; 2021.
- Ho KM, Pavey W. Applying the cell-based coagulation model in the management of critical bleeding. Anaesth Intensive Care. Mar 2017;45(2):166-176. doi:10.1177/0310057x1704500206
- Chernysh IN, Nagaswami C, Kosolapova S, et al. The distinctive structure and composition of arterial and venous thrombi and pulmonary emboli. Sci Rep. Mar 20 2020;10(1):5112. doi:10.1038/s41598-020-59526-x
- Badireddy M, Mudipalli VR. Deep Venous Thrombosis Prophylaxis. StatPearls. 2021.
- Ageno W, Beyer-Westendorf J, Garcia DA, Lazo-Langner A, McBane RD, Paciaroni M. Guidance for the management of venous thrombosis in unusual sites. Journal of thrombosis and thrombolysis. Jan 2016;41(1):129-43. doi:10.1007/s11239-015-1308-1
- Chan NC, Weitz JI. Recent advances in understanding, diagnosing and treating venous thrombosis. F1000Research. 2020;9doi:10.12688/f1000research.27115.1
- Metz AK, Diaz JA, Obi AT, Wakefield TW, Myers DD, Henke PK. Venous Thrombosis and Post-Thrombotic Syndrome: From Novel Biomarkers to Biology. Methodist DeBakey cardiovascular journal. Jul-Sep 2018;14(3):173-181. doi:10.14797/mdcj-14-3-173
- Winter MP, Schernthaner GH, Lang IM. Chronic complications of venous thromboembolism. J Thromb Haemost. Aug 2017;15(8):1531-1540. doi:10.1111/jth.13741
- Prevention; CfDCa. Data and Statistics on Venous Thromboembolism. Accessed September 3, 2021, https://www.cdc.gov/ncbddd/dvt/data.html
- Chang WT, Chang CL, Ho CH, Hong CS, Wang JJ, Chen ZC. Long‐Term Effects of Unprovoked Venous Thromboembolism on Mortality and Major Cardiovascular Events. Journal of the American Heart Association. 6(5):e005466. doi:10.1161/JAHA.117.005466
- Di Minno MN, Ambrosino P, Ambrosini F, Tremoli E, Di Minno G, Dentali F. Prevalence of deep vein thrombosis and pulmonary embolism in patients with superficial vein thrombosis: a systematic review and meta-analysis. J Thromb Haemost. May 2016;14(5):964-72. doi:10.1111/jth.13279
- Lebas H, Yahiaoui K, Martos R, Boulaftali Y. Platelets Are at the Nexus of Vascular Diseases. Front Cardiovasc Med. 2019;6:132. doi:10.3389/fcvm.2019.00132
- De Angelis G, Cimon K, Sinclair A, et al. CADTH Optimal Use Reports. Monitoring for Atrial Fibrillation in Discharged Stroke and Transient Ischemic Attack Patients: Recommendations . Canadian Agency for Drugs and Technologies in Health Copyright © CADTH 2016.; 2016.
- Oladiran O, Nwosu I. Stroke risk stratification in atrial fibrillation: a review of common risk factors. J Community Hosp Intern Med Perspect. Apr 2019;9(2):113-120. doi:10.1080/20009666.2019.1593781
- Lyaker MR, Tulman DB, Dimitrova GT, Pin RH, Papadimos TJ. Arterial embolism. Int J Crit Illn Inj Sci. Jan 2013;3(1):77-87. doi:10.4103/2229-5151.109429
- Slíva J, Charalambous C, Bultas J, Karetová D. A new strategy for the treatment of atherothrombosis - inhibition of inflammation. Physiological research / Academia Scientiarum Bohemoslovaca. Nov 22 2019;68(Suppl 1):S17-s30. doi:10.33549/physiolres.934327
- Fioranelli M, Bottaccioli AG, Bottaccioli F, Bianchi M, Rovesti M, Roccia MG. Stress and Inflammation in Coronary Artery Disease: A Review Psychoneuroendocrineimmunology-Based. Front Immunol. 2018;9:2031. doi:10.3389/fimmu.2018.02031
- Chen Y, Ju LA. Biomechanical thrombosis: the dark side of force and dawn of mechano-medicine. Stroke Vasc Neurol. Jun 2020;5(2):185-197. doi:10.1136/svn-2019-000302
- Shah PK, Lecis D. Inflammation in atherosclerotic cardiovascular disease. F1000Research. 2019;8doi:10.12688/f1000research.18901.1
- Ketelhuth DFJ, Lutgens E, Bäck M, et al. Immunometabolism and atherosclerosis: perspectives and clinical significance: a position paper from the Working Group on Atherosclerosis and Vascular Biology of the European Society of Cardiology. Cardiovasc Res. Jul 1 2019;115(9):1385-1392. doi:10.1093/cvr/cvz166
- Gross PL, Chan NC. Thromboembolism in Older Adults. Front Med (Lausanne). 2020;7:470016. doi:10.3389/fmed.2020.470016
- Folsom AR, Cushman M. Exploring Opportunities for Primary Prevention of Unprovoked Venous Thromboembolism: Ready for Prime Time? J Am Heart Assoc. Dec 2020;9(23):e019395. doi:10.1161/jaha.120.019395
- Detopoulou P, Demopoulos CA, Antonopoulou S. Micronutrients, Phytochemicals and Mediterranean Diet: A Potential Protective Role against COVID-19 through Modulation of PAF Actions and Metabolism. Nutrients. Jan 30 2021;13(2)doi:10.3390/nu13020462
- Tsoupras A, Lordan R, Zabetakis I. Thrombosis and COVID-19: The Potential Role of Nutrition. Frontiers in nutrition. 2020;7:583080. doi:10.3389/fnut.2020.583080
- Soliman GA. Dietary Fiber, Atherosclerosis, and Cardiovascular Disease. Nutrients. May 23 2019;11(5)doi:10.3390/nu11051155
- Violi F, Pastori D, Pignatelli P, Carnevale R. Nutrition, Thrombosis, and Cardiovascular Disease. Circ Res. May 8 2020;126(10):1415-1442. doi:10.1161/circresaha.120.315892
- Magrone T, Russo MA, Jirillo E. Cocoa and Dark Chocolate Polyphenols: From Biology to Clinical Applications. Front Immunol. 2017;8:677. doi:10.3389/fimmu.2017.00677
- Aprotosoaie AC, Miron A, Trifan A, Luca VS, Costache, II. The Cardiovascular Effects of Cocoa Polyphenols-An Overview. Diseases. Dec 17 2016;4(4)doi:10.3390/diseases4040039
- Garcia JP, Santana A, Baruqui DL, Suraci N. The Cardiovascular effects of chocolate. Reviews in cardiovascular medicine. Dec 30 2018;19(4):123-127. doi:10.31083/j.rcm.2018.04.3187
- Pearson DA, Paglieroni TG, Rein D, et al. The effects of flavanol-rich cocoa and aspirin on ex vivo platelet function. Thromb Res. May 15 2002;106(4-5):191-7. doi:10.1016/s0049-3848(02)00128-7
- Ko EY, Nile SH, Jung YS, Keum YS. Antioxidant and antiplatelet potential of different methanol fractions and flavonols extracted from onion (Allium cepa L.). 3 Biotech. Mar 2018;8(3):155. doi:10.1007/s13205-018-1184-4
- Bahadoran Z, Mirmiran P, Momenan AA, Azizi F. Allium vegetable intakes and the incidence of cardiovascular disease, hypertension, chronic kidney disease, and type 2 diabetes in adults: a longitudinal follow-up study. Journal of hypertension. Sep 2017;35(9):1909-1916. doi:10.1097/hjh.0000000000001356
- Hubbard GP, Wolffram S, de Vos R, Bovy A, Gibbins JM, Lovegrove JA. Ingestion of onion soup high in quercetin inhibits platelet aggregation and essential components of the collagen-stimulated platelet activation pathway in man: a pilot study. The British journal of nutrition. Sep 2006;96(3):482-8.
- Ansary J, Forbes-Hernández TY, Gil E, et al. Potential Health Benefit of Garlic Based on Human Intervention Studies: A Brief Overview. Antioxidants (Basel, Switzerland). Jul 15 2020;9(7)doi:10.3390/antiox9070619
- Ali M, Thomson M. Consumption of a garlic clove a day could be beneficial in preventing thrombosis. Prostaglandins Leukot Essent Fatty Acids . Sep 1995;53(3):211-2. doi:10.1016/0952-3278(95)90118-3
- Li C, Li J, Jiang F, et al. Vasculoprotective effects of ginger (Zingiber officinale Roscoe) and underlying molecular mechanisms. Food Funct . Mar 15 2021;12(5):1897-1913. doi:10.1039/d0fo02210a
- Fakhri S, Patra JK, Das SK, Das G, Majnooni MB, Farzaei MH. Ginger and Heart Health: From Mechanisms to Therapeutics. Current molecular pharmacology. Dec 8 2020;doi:10.2174/1874467213666201209105005
- Marx W, McKavanagh D, McCarthy AL, et al. The Effect of Ginger (Zingiber officinale) on Platelet Aggregation: A Systematic Literature Review. PLoS One. 2015;10(10):e0141119. doi:10.1371/journal.pone.0141119
- Wang Y, Yu H, Zhang X, et al. Evaluation of daily ginger consumption for the prevention of chronic diseases in adults: A cross-sectional study. Nutrition (Burbank, Los Angeles County, Calif). Apr 2017;36:79-84. doi:10.1016/j.nut.2016.05.009
- Ali MY, Sina AA, Khandker SS, et al. Nutritional Composition and Bioactive Compounds in Tomatoes and Their Impact on Human Health and Disease: A Review. Foods. Dec 26 2020;10(1)doi:10.3390/foods10010045
- Fielding JM, Rowley KG, Cooper P, K OD. Increases in plasma lycopene concentration after consumption of tomatoes cooked with olive oil. Asia Pac J Clin Nutr. 2005;14(2):131-6.
- Mazidi M, Katsiki N, George ES, Banach M. Tomato and lycopene consumption is inversely associated with total and cause-specific mortality: a population-based cohort study, on behalf of the International Lipid Expert Panel (ILEP). The British journal of nutrition. Dec 28 2020;124(12):1303-1310. doi:10.1017/s0007114519002150
- Cheng HM, Koutsidis G, Lodge JK, Ashor AW, Siervo M, Lara J. Lycopene and tomato and risk of cardiovascular diseases: A systematic review and meta-analysis of epidemiological evidence. Crit Rev Food Sci Nutr. 2019;59(1):141-158. doi:10.1080/10408398.2017.1362630
- Olas B. A review of in vitro studies of the anti-platelet potential of citrus fruit flavonoids. Food and chemical toxicology : an international journal published for the British Industrial Biological Research Association . Apr 2021;150:112090. doi:10.1016/j.fct.2021.112090
- Hyson DA. A review and critical analysis of the scientific literature related to 100% fruit juice and human health. Adv Nutr. Jan 2015;6(1):37-51. doi:10.3945/an.114.005728
- Petsini F, Fragopoulou E, Antonopoulou S. Fish consumption and cardiovascular disease related biomarkers: A review of clinical trials. Crit Rev Food Sci Nutr. 2019;59(13):2061-2071. doi:10.1080/10408398.2018.1437388
- Burr ML, Fehily AM, Gilbert JF, et al. Effects of changes in fat, fish, and fibre intakes on death and myocardial reinfarction: diet and reinfarction trial (DART). Lancet. Sep 30 1989;2(8666):757-61. doi:10.1016/s0140-6736(89)90828-3
- Isaksen T, Evensen LH, Brækkan SK, Hansen JB. Dietary Intake of Marine Polyunsaturated n-3 Fatty Acids and Risk of Recurrent Venous Thromboembolism. Thrombosis and haemostasis. Dec 2019;119(12):2053-2063. doi:10.1055/s-0039-1697663
- Golanski J, Szymanska P, Rozalski M. Effects of Omega-3 Polyunsaturated Fatty Acids and Their Metabolites on Haemostasis-Current Perspectives in Cardiovascular Disease. International journal of molecular sciences. Feb 27 2021;22(5)doi:10.3390/ijms22052394
- Liput KP, Lepczyński A, Ogłuszka M, et al. Effects of Dietary n-3 and n-6 Polyunsaturated Fatty Acids in Inflammation and Cancerogenesis. International journal of molecular sciences. Jun 28 2021;22(13)doi:10.3390/ijms22136965
- Katsiki N, Pérez-Martínez P, Lopez-Miranda J. Olive Oil Intake and Cardiovascular Disease Prevention: "Seek and You Shall Find". Current cardiology reports. May 7 2021;23(6):64. doi:10.1007/s11886-021-01496-1
- Capurso C, Massaro M, Scoditti E, Vendemiale G, Capurso A. Vascular effects of the Mediterranean diet part I: anti-hypertensive and anti-thrombotic effects. Vascul Pharmacol. Dec 2014;63(3):118-26. doi:10.1016/j.vph.2014.10.001
- Widmer RJ, Freund MA, Flammer AJ, et al. Beneficial effects of polyphenol-rich olive oil in patients with early atherosclerosis. European journal of nutrition. Apr 2013;52(3):1223-31. doi:10.1007/s00394-012-0433-2
- Fragopoulou E, Antonopoulou S. The French paradox three decades later: Role of inflammation and thrombosis. Clin Chim Acta. Nov 2020;510:160-169. doi:10.1016/j.cca.2020.07.013
- Liberale L, Bonaventura A, Montecucco F, Dallegri F, Carbone F. Impact of Red Wine Consumption on Cardiovascular Health. Curr Med Chem. 2019;26(19):3542-3566. doi:10.2174/0929867324666170518100606
- Larsson SC, Wallin A, Wolk A, Markus HS. Differing association of alcohol consumption with different stroke types: a systematic review and meta-analysis. BMC Med. Nov 24 2016;14(1):178. doi:10.1186/s12916-016-0721-4
- USDHHS. US Department of Health and Human Services. 2015–2020 Dietary Guidelines. Available at https://health.gov/our-work/food-nutrition/previous-dietary-guidelines/2015 . Last updated 12/29/20. Accessed 08/18/21. 2020;
- Panchal G, Mahmood M, Lip GYH. Revisiting the risks of incident atrial fibrillation: a narrative review. Part 2. Kardiologia polska. May 24 2019;77(5):515-524. doi:10.33963/kp.14846
- Lippi G, Mattiuzzi C, Franchini M. Alcohol consumption and venous thromboembolism: friend or foe? Intern Emerg Med. Dec 2015;10(8):907-13. doi:10.1007/s11739-015-1327-0
- Johansson M, Johansson L, Wennberg M, Lind M. Alcohol consumption and risk of first-time venous thromboembolism in men and women. Thrombosis and haemostasis. 2019;119(06):962-970.
- Chen M, Ji M, Chen T, Hong X, Jia Y. Alcohol consumption and risk for venous thromboembolism: a meta-analysis of prospective studies. Frontiers in nutrition. 2020;7:32.
- Hanna K, Khalid A, Hamidi M, et al. Chronic alcohol consumption and risk of deep venous thrombosis: a propensity-matched analysis. Journal of Surgical Research. 2019;244:251-256.
- Di Minno A, Frigerio B, Spadarella G, et al. Old and new oral anticoagulants: Food, herbal medicines and drug interactions. Blood reviews. Jul 2017;31(4):193-203. doi:10.1016/j.blre.2017.02.001
- NIH. National Institutes of Health: Office of Dietary Supplements. Vitamin K Fact Sheet for Health Professionals. Available at https://ods.od.nih.gov/factsheets/VitaminK-HealthProfessional/ . Last update 03/29/2021. Accessed 08/18/2021. 2021;
- Violi F, Lip GY, Pignatelli P, Pastori D. Interaction Between Dietary Vitamin K Intake and Anticoagulation by Vitamin K Antagonists: Is It Really True?: A Systematic Review. Medicine. Mar 2016;95(10):e2895. doi:10.1097/md.0000000000002895
- Olsen LN, Fischer M, Evans PA, Gliemann L, Hellsten Y. Does Exercise Influence the Susceptibility to Arterial Thrombosis? An Integrative Perspective. Front Physiol. 2021;12:636027. doi:10.3389/fphys.2021.636027
- Gronek P, Wielinski D, Cyganski P, et al. A Review of Exercise as Medicine in Cardiovascular Disease: Pathology and Mechanism. Aging Dis. Apr 2020;11(2):327-340. doi:10.14336/ad.2019.0516
- Chung MK, Eckhardt LL, Chen LY, et al. Lifestyle and Risk Factor Modification for Reduction of Atrial Fibrillation: A Scientific Statement From the American Heart Association. Circulation. Apr 21 2020;141(16):e750-e772. doi:10.1161/cir.0000000000000748
- Heber S, Fischer B, Sallaberger-Lehner M, et al. Effects of high-intensity interval training on platelet function in cardiac rehabilitation: a randomised controlled trial. Heart. Jan 2020;106(1):69-79. doi:10.1136/heartjnl-2019-315130
- Evensen LH, Brækkan SK, Hansen JB. Regular Physical Activity and Risk of Venous Thromboembolism. Semin Thromb Hemost. Nov 2018;44(8):765-779. doi:10.1055/s-0038-1673636
- Guo M, Lu L, Sun Y, Li L, Wu M, Lang J. Comprehensive functional exercises with patient education for the prevention of venous thrombosis after major gynecologic surgery: A randomized controlled study. Thromb Res. Jun 2019;178:69-74. doi:10.1016/j.thromres.2019.04.013
- Liu K, Zhou Y, Xie W, et al. Handgrip exercise reduces peripherally-inserted central catheter-related venous thrombosis in patients with solid cancers: A randomized controlled trial. International journal of nursing studies. Oct 2018;86:99-106. doi:10.1016/j.ijnurstu.2018.06.004
- Wang Z, Chen Q, Ye M, Shi GH, Zhang B. Active Ankle Movement May Prevent Deep Vein Thrombosis in Patients Undergoing Lower Limb Surgery. Annals of vascular surgery. Apr 2016;32:65-72. doi:10.1016/j.avsg.2015.10.012
- Sandrini L, Ieraci A, Amadio P, Zarà M, Barbieri SS. Impact of Acute and Chronic Stress on Thrombosis in Healthy Individuals and Cardiovascular Disease Patients. International journal of molecular sciences. Oct 22 2020;21(21)doi:10.3390/ijms21217818
- von Känel R. Acute mental stress and hemostasis: When physiology becomes vascular harm. Thromb Res. Feb 2015;135 Suppl 1:S52-5. doi:10.1016/s0049-3848(15)50444-1
- Krittanawong C, Kumar A, Wang Z, et al. Meditation and Cardiovascular Health in the US. The American journal of cardiology. Sep 15 2020;131:23-26. doi:10.1016/j.amjcard.2020.06.043
- Nijjar PS, Connett JE, Lindquist R, et al. Randomized Trial of Mindfulness-Based Stress Reduction in Cardiac Patients Eligible for Cardiac Rehabilitation. Sci Rep. Dec 5 2019;9(1):18415. doi:10.1038/s41598-019-54932-2
- Fan H, Zhou J, Yuan Z. Meta-Analysis Comparing the Effect of Combined Omega-3 + Statin Therapy Versus Statin Therapy Alone on Coronary Artery Plaques. The American journal of cardiology. May 25 2021;doi:10.1016/j.amjcard.2021.04.013
- Bernasconi AA, Wiest MM, Lavie CJ, Milani RV, Laukkanen JA. Effect of Omega-3 Dosage on Cardiovascular Outcomes: An Updated Meta-Analysis and Meta-Regression of Interventional Trials. Mayo Clin Proc. Feb 2021;96(2):304-313. doi:10.1016/j.mayocp.2020.08.034
- Saber H, Yakoob MY, Shi P, et al. Omega-3 Fatty Acids and Incident Ischemic Stroke and Its Atherothrombotic and Cardioembolic Subtypes in 3 US Cohorts. Stroke. Oct 2017;48(10):2678-2685. doi:10.1161/strokeaha.117.018235
- Lombardi M, Chiabrando JG, Vescovo GM, et al. Impact of Different Doses of Omega-3 Fatty Acids on Cardiovascular Outcomes: a Pairwise and Network Meta-analysis. Curr Atheroscler Rep. Jul 16 2020;22(9):45. doi:10.1007/s11883-020-00865-5
- Bhatt DL, Steg PG, Miller M, et al. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. The New England journal of medicine. Jan 3 2019;380(1):11-22. doi:10.1056/NEJMoa1812792
- Kapoor K, Alfaddagh A, Al Rifai M, et al. Association Between Omega-3 Fatty Acid Levels and Risk for Incident Major Bleeding Events and Atrial Fibrillation: MESA. J Am Heart Assoc. Jun 2021;10(11):e021431. doi:10.1161/jaha.121.021431
- Ramadeen A, Connelly KA, Leong-Poi H, et al. Docosahexaenoic acid, but not eicosapentaenoic acid, supplementation reduces vulnerability to atrial fibrillation. Circ Arrhythm Electrophysiol. Oct 2012;5(5):978-83. doi:10.1161/circep.112.971515
- Zheng X, Jia R, Li Y, Liu T, Wang Z. Omega-3 fatty acids reduce post-operative risk of deep vein thrombosis and pulmonary embolism after surgery for elderly patients with proximal femoral fractures: a randomized placebo-controlled, double-blind clinical trial. Int Orthop. Oct 2020;44(10):2089-2093. doi:10.1007/s00264-020-04610-0
- Viecelli AK, Polkinghorne KR, Pascoe EM, et al. Fish oil and aspirin effects on arteriovenous fistula function: Secondary outcomes of the randomised omega-3 fatty acids (Fish oils) and Aspirin in Vascular access OUtcomes in REnal Disease (FAVOURED) trial. PLoS One. 2019;14(3):e0213274. doi:10.1371/journal.pone.0213274
- Isaksen T, Evensen LH, Johnsen SH, et al. Dietary intake of marine n-3 polyunsaturated fatty acids and future risk of venous thromboembolism. Research and practice in thrombosis and haemostasis. Jan 2019;3(1):59-69. doi:10.1002/rth2.12168
- Reiner MF, Stivala S, Limacher A, et al. Omega-3 fatty acids predict recurrent venous thromboembolism or total mortality in elderly patients with acute venous thromboembolism. J Thromb Haemost. Jan 2017;15(1):47-56. doi:10.1111/jth.13553
- Zuo W, Yan F, Zhang B, Li J, Mei D. Advances in the Studies of Ginkgo Biloba Leaves Extract on Aging-Related Diseases. Aging Dis. Dec 2017;8(6):812-826. doi:10.14336/ad.2017.0615
- Wu Y, Li S, Cui W, Zu X, Du J, Wang F. Ginkgo biloba extract improves coronary blood flow in healthy elderly adults: role of endothelium-dependent vasodilation. Phytomedicine. Mar 2008;15(3):164-9. doi:10.1016/j.phymed.2007.12.002
- Wu Y, Li S, Cui W, Zu X, Wang F, Du J. Ginkgo biloba extract improves coronary blood flow in patients with coronary artery disease: role of endothelium-dependent vasodilation. Planta Med. Jun 2007;73(7):624-8. doi:10.1055/s-2007-981536
- Jung F, Mrowietz C, Kiesewetter H, Wenzel E. Effect of Ginkgo biloba on fluidity of blood and peripheral microcirculation in volunteers. Arzneimittel-Forschung. May 1990;40(5):589-93.
- Sarkar C, Quispe C, Jamaddar S, et al. Therapeutic promises of ginkgolide A: A literature-based review. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie . Dec 2020;132:110908. doi:10.1016/j.biopha.2020.110908
- Li X, Lu L, Chen J, Zhang C, Chen H, Huang H. New Insight into the Mechanisms of Ginkgo Biloba Extract in Vascular Aging Prevention. Curr Vasc Pharmacol. 2020;18(4):334-345. doi:10.2174/1570161117666190621150725
- Janssens D, Michiels C, Guillaume G, Cuisinier B, Louagie Y, Remacle J. Increase in circulating endothelial cells in patients with primary chronic venous insufficiency: protective effect of Ginkor Fort in a randomized double-blind, placebo-controlled clinical trial. Journal of cardiovascular pharmacology. Jan 1999;33(1):7-11. doi:10.1097/00005344-199901000-00002
- Eisvand F, Razavi BM, Hosseinzadeh H. The effects of Ginkgo biloba on metabolic syndrome: A review. Phytother Res. Aug 2020;34(8):1798-1811. doi:10.1002/ptr.6646
- Tian J, Liu Y, Chen K. Ginkgo biloba Extract in Vascular Protection: Molecular Mechanisms and Clinical Applications. Curr Vasc Pharmacol. 2017;15(6):532-548. doi:10.2174/1570161115666170713095545
- Chiu YL, Tsai WC, Wu CH, et al. Ginkgo biloba Induces Thrombomodulin Expression and Tissue-Type Plasminogen Activator Secretion via the Activation of Krüppel-Like Factor 2 within Endothelial Cells. The American journal of Chinese medicine. 2020;48(2):357-372. doi:10.1142/s0192415x20500184
- Chen TR, Wei LH, Guan XQ, et al. Biflavones from Ginkgo biloba as inhibitors of human thrombin. Bioorganic chemistry. Nov 2019;92:103199. doi:10.1016/j.bioorg.2019.103199
- Kellermann AJ, Kloft C. Is there a risk of bleeding associated with standardized Ginkgo biloba extract therapy? A systematic review and meta-analysis. Pharmacotherapy. May 2011;31(5):490-502. doi:10.1592/phco.31.5.490
- Ryu KH, Han HY, Lee SY, et al. Ginkgo biloba extract enhances antiplatelet and antithrombotic effects of cilostazol without prolongation of bleeding time. Thromb Res. Jul 2009;124(3):328-34. doi:10.1016/j.thromres.2009.02.010
- Sobenin IA, Myasoedova VA, Iltchuk MI, Zhang DW, Orekhov AN. Therapeutic effects of garlic in cardiovascular atherosclerotic disease. Chinese journal of natural medicines. Oct 2019;17(10):721-728. doi:10.1016/s1875-5364(19)30088-3
- Olas B. Anti-Aggregatory Potential of Selected Vegetables-Promising Dietary Components for the Prevention and Treatment of Cardiovascular Disease. Adv Nutr. Mar 1 2019;10(2):280-290. doi:10.1093/advances/nmy085
- Kim L, Lim Y, Park SY, et al. A comparative study of the antithrombotic effect through activated endothelium of garlic powder and tomato extracts using a rodent model of collagen and epinephrine induced thrombosis. Food Sci Biotechnol. Oct 2018;27(5):1513-1518. doi:10.1007/s10068-018-0469-z
- el-Sabban F, Radwan GM. Influence of garlic compared to aspirin on induced photothrombosis in mouse pial microvessels, in vivo. Thromb Res. Oct 15 1997;88(2):193-203. doi:10.1016/s0049-3848(97)00230-2
- Apitz-Castro R, Badimon JJ, Badimon L. A garlic derivative, ajoene, inhibits platelet deposition on severely damaged vessel wall in an in vivo porcine experimental model. Thromb Res. Aug 1 1994;75(3):243-9. doi:10.1016/0049-3848(94)90235-6
- Apitz-Castro R, Badimon JJ, Badimon L. Effect of ajoene, the major antiplatelet compound from garlic, on platelet thrombus formation. Thromb Res. Oct 15 1992;68(2):145-55. doi:10.1016/0049-3848(92)90030-e
- Fakhar H, Hashemi Tayer A. Effect of the Garlic Pill in comparison with Plavix on Platelet Aggregation and Bleeding Time. Iran J Ped Hematol Oncol. 2012;2(4):146-52.
- Macan H, Uykimpang R, Alconcel M, et al. Aged garlic extract may be safe for patients on warfarin therapy. J Nutr. Mar 2006;136(3 Suppl):793s-795s. doi:10.1093/jn/136.3.793S
- Davinelli S, Corbi G, Zarrelli A, et al. Short-term supplementation with flavanol-rich cocoa improves lipid profile, antioxidant status and positively influences the AA/EPA ratio in healthy subjects. J Nutr Biochem. Nov 2018;61:33-39. doi:10.1016/j.jnutbio.2018.07.011
- Tzounis X, Rodriguez-Mateos A, Vulevic J, Gibson GR, Kwik-Uribe C, Spencer JP. Prebiotic evaluation of cocoa-derived flavanols in healthy humans by using a randomized, controlled, double-blind, crossover intervention study. Am J Clin Nutr. Jan 2011;93(1):62-72. doi:10.3945/ajcn.110.000075
- Montagnana M, Danese E, Angelino D, et al. Dark chocolate modulates platelet function with a mechanism mediated by flavan-3-ol metabolites. Medicine. Dec 2018;97(49):e13432. doi:10.1097/md.0000000000013432
- Montagnana M, Danese E, Salvagno GL, Lippi G. Short-term effect of dark chocolate consumption on routine haemostasis testing. International journal of food sciences and nutrition. Aug 2017;68(5):613-616. doi:10.1080/09637486.2016.1268101
- von Känel R, Meister RE, Stutz M, et al. Effects of dark chocolate consumption on the prothrombotic response to acute psychosocial stress in healthy men. Thrombosis and haemostasis. Dec 2014;112(6):1151-8. doi:10.1160/th14-05-0450
- Sansone R, Rodriguez-Mateos A, Heuel J, et al. Cocoa flavanol intake improves endothelial function and Framingham Risk Score in healthy men and women: a randomised, controlled, double-masked trial: the Flaviola Health Study. The British journal of nutrition. Oct 28 2015;114(8):1246-55. doi:10.1017/s0007114515002822
- Horn P, Amabile N, Angeli FS, et al. Dietary flavanol intervention lowers the levels of endothelial microparticles in coronary artery disease patients. The British journal of nutrition. Apr 14 2014;111(7):1245-52. doi:10.1017/s0007114513003693
- Kim K, Brothers RM. Acute consumption of flavanol-rich cocoa beverage improves attenuated cutaneous microvascular function in healthy young African Americans. Microvascular research. Mar 2020;128:103931. doi:10.1016/j.mvr.2019.103931
- Pereira T, Bergqvist J, Vieira C, Grüner Sveälv B, Castanheira J, Conde J. Randomized study of the effects of cocoa-rich chocolate on the ventricle-arterial coupling and vascular function of young, healthy adults. Nutrition (Burbank, Los Angeles County, Calif). Jul-Aug 2019;63-64:175-183. doi:10.1016/j.nut.2019.02.017
- Okamoto T, Kobayashi R, Natsume M, Nakazato K. Habitual cocoa intake reduces arterial stiffness in postmenopausal women regardless of intake frequency: a randomized parallel-group study. Clin Interv Aging. 2016;11:1645-1652. doi:10.2147/cia.S118152
- Rassaf T, Rammos C, Hendgen-Cotta UB, et al. Vasculoprotective Effects of Dietary Cocoa Flavanols in Patients on Hemodialysis: A Double-Blind, Randomized, Placebo-Controlled Trial. Clinical journal of the American Society of Nephrology : CJASN. Jan 7 2016;11(1):108-18. doi:10.2215/cjn.05560515
- Rull G, Mohd-Zain ZN, Shiel J, et al. Effects of high flavanol dark chocolate on cardiovascular function and platelet aggregation. Vascul Pharmacol. Aug 2015;71:70-8. doi:10.1016/j.vph.2015.02.010
- Heiss C, Sansone R, Karimi H, et al. Impact of cocoa flavanol intake on age-dependent vascular stiffness in healthy men: a randomized, controlled, double-masked trial. Age (Dordr). Jun 2015;37(3):9794. doi:10.1007/s11357-015-9794-9
- Shimizu M, Miyazaki T, Takagi A, et al. Low coenzyme Q10 levels in patients with acute cardiovascular disease are associated with long-term mortality. Heart Vessels. Mar 2021;36(3):401-407. doi:10.1007/s00380-020-01698-7
- Martelli A, Testai L, Colletti A, Cicero AFG. Coenzyme Q(10): Clinical Applications in Cardiovascular Diseases. Antioxidants (Basel, Switzerland). Apr 22 2020;9(4)doi:10.3390/antiox9040341
- Rabanal-Ruiz Y, Llanos-González E, Alcain FJ. The Use of Coenzyme Q10 in Cardiovascular Diseases. Antioxidants (Basel, Switzerland). May 10 2021;10(5)doi:10.3390/antiox10050755
- Arenas-Jal M, Suñé-Negre JM, García-Montoya E. Coenzyme Q10 supplementation: Efficacy, safety, and formulation challenges. Compr Rev Food Sci Food Saf. Mar 2020;19(2):574-594. doi:10.1111/1541-4337.12539
- Ya F, Xu XR, Shi Y, et al. Coenzyme Q10 Upregulates Platelet cAMP/PKA Pathway and Attenuates Integrin αIIbβ3 Signaling and Thrombus Growth. Mol Nutr Food Res. Dec 2019;63(23):e1900662. doi:10.1002/mnfr.201900662
- Raizner AE, Quiñones MA. Coenzyme Q(10) for Patients With Cardiovascular Disease: JACC Focus Seminar. Journal of the American College of Cardiology. Feb 9 2021;77(5):609-619. doi:10.1016/j.jacc.2020.12.009
- Alehagen U, Aaseth J, Lindahl TL, Larsson A, Alexander J. Dietary Supplementation with Selenium and Coenzyme Q(10) Prevents Increase in Plasma D-Dimer While Lowering Cardiovascular Mortality in an Elderly Swedish Population. Nutrients. Apr 17 2021;13(4)doi:10.3390/nu13041344
- Alehagen U, Alexander J, Aaseth J, Larsson A, Lindahl TL. Significant decrease of von Willebrand factor and plasminogen activator inhibitor-1 by providing supplementation with selenium and coenzyme Q10 to an elderly population with a low selenium status. European journal of nutrition. 2020/12/01 2020;59(8):3581-3590. doi:10.1007/s00394-020-02193-5
- Sabbatinelli J, Orlando P, Galeazzi R, et al. Ubiquinol Ameliorates Endothelial Dysfunction in Subjects with Mild-to-Moderate Dyslipidemia: A Randomized Clinical Trial. Nutrients. Apr 15 2020;12(4)doi:10.3390/nu12041098
- Pérez-Sánchez C, Aguirre M, Ruiz-Limón P, et al. Ubiquinol Effects on Antiphospholipid Syndrome Prothrombotic Profile: A Randomized, Placebo-Controlled Trial. Arteriosclerosis, thrombosis, and vascular biology. Oct 2017;37(10):1923-1932. doi:10.1161/atvbaha.117.309225
- Gulati OP. Pycnogenol® in chronic venous insufficiency and related venous disorders. Phytother Res. Mar 2014;28(3):348-62. doi:10.1002/ptr.5019
- Belcaro G, Cornelli U, Dugall M, Hosoi M, Cotellese R, Feragalli B. Long-haul flights, edema, and thrombotic events: prevention with stockings and Pycnogenol® supplementation (LONFLIT Registry Study). Minerva cardioangiologica. Apr 2018;66(2):152-159. doi:10.23736/s0026-4725.17.04577-7
- Belcaro G, Cesarone MR, Rohdewald P, et al. Prevention of venous thrombosis and thrombophlebitis in long-haul flights with pycnogenol. Clinical and applied thrombosis/hemostasis : official journal of the International Academy of Clinical and Applied Thrombosis/Hemostasis . Oct 2004;10(4):373-7.
- Errichi BM, Belcaro G, Hosoi M, et al. Prevention of post thrombotic syndrome with Pycnogenol® in a twelve month study. Panminerva medica. Sep 2011;53(3 Suppl 1):21-7.
- Belcaro G, Dugall M, Hu S, et al. Prevention of recurrent venous thrombosis and post-thrombotic syndrome. Minerva cardioangiologica. Jun 2018;66(3):238-245. doi:10.23736/S0026-4725.18.04618-2
- Nishioka K, Hidaka T, Nakamura S, et al. Pycnogenol, French maritime pine bark extract, augments endothelium-dependent vasodilation in humans. Hypertension research : official journal of the Japanese Society of Hypertension . Sep 2007;30(9):775-80. doi:10.1291/hypres.30.775
- Enseleit F, Sudano I, Periat D, et al. Effects of Pycnogenol on endothelial function in patients with stable coronary artery disease: a double-blind, randomized, placebo-controlled, cross-over study. Eur Heart J. Jul 2012;33(13):1589-97. doi:10.1093/eurheartj/ehr482
- Araghi-Niknam M, Hosseini S, Larson D, Rohdewald P, Watson RR. Pine bark extract reduces platelet aggregation. Integrative medicine : integrating conventional and alternative medicine . Mar 21 2000;2(2):73-77.
- Belcaro G, Dugall M, Bradford HD, et al. Recurrent retinal vein thrombosis: prevention with Aspirin, Pycnogenol®, ticlopidine, or sulodexide. Minerva cardioangiologica. Apr 2019;67(2):109-114. doi:10.23736/s0026-4725.19.04891-6
- Rodriguez P, Belcaro G, Dugall M, et al. Recurrence of retinal vein thrombosis with Pycnogenol® or Aspirin® supplementation: a registry study. Panminerva medica. Sep 2015;57(3):121-5.
- Lässiger-Herfurth A, Pontarollo G, Grill A, Reinhardt C. The Gut Microbiota in Cardiovascular Disease and Arterial Thrombosis. Microorganisms. Dec 13 2019;7(12)doi:10.3390/microorganisms7120691
- Lippi G, Danese E, Mattiuzzi C, Favaloro EJ. The Intriguing Link between the Intestinal Microbiota and Cardiovascular Disease. Semin Thromb Hemost. Sep 2017;43(6):609-613. doi:10.1055/s-0036-1597903
- Wu Y, Zhang Q, Ren Y, Ruan Z. Effect of probiotic Lactobacillus on lipid profile: A systematic review and meta-analysis of randomized, controlled trials. PLoS One. 2017;12(6):e0178868. doi:10.1371/journal.pone.0178868
- Jones ML, Martoni CJ, Prakash S. Cholesterol lowering and inhibition of sterol absorption by Lactobacillus reuteri NCIMB 30242: a randomized controlled trial. Eur J Clin Nutr. Nov 2012;66(11):1234-41. doi:10.1038/ejcn.2012.126
- Jones ML, Martoni CJ, Prakash S. Oral supplementation with probiotic L. reuteri NCIMB 30242 increases mean circulating 25-hydroxyvitamin D: a post hoc analysis of a randomized controlled trial. J Clin Endocrinol Metab. Jul 2013;98(7):2944-51. doi:10.1210/jc.2012-4262
- Jones ML, Martoni CJ, Parent M, Prakash S. Cholesterol-lowering efficacy of a microencapsulated bile salt hydrolase-active Lactobacillus reuteri NCIMB 30242 yoghurt formulation in hypercholesterolaemic adults. Br J Nutr. May 2012;107(10):1505-13. doi:10.1017/s0007114511004703
- Matsumoto M, Kitada Y, Naito Y. Endothelial Function is improved by Inducing Microbial Polyamine Production in the Gut: A Randomized Placebo-Controlled Trial. Nutrients. May 27 2019;11(5)doi:10.3390/nu11051188
- Naruszewicz M, Johansson ML, Zapolska-Downar D, Bukowska H. Effect of Lactobacillus plantarum 299v on cardiovascular disease risk factors in smokers. Am J Clin Nutr. Dec 2002;76(6):1249-55. doi:10.1093/ajcn/76.6.1249
- Stiksrud B, Nowak P, Nwosu FC, et al. Reduced Levels of D-dimer and Changes in Gut Microbiota Composition After Probiotic Intervention in HIV-Infected Individuals on Stable ART. J Acquir Immune Defic Syndr. Dec 1 2015;70(4):329-37. doi:10.1097/qai.0000000000000784
- Zhang X, Tong Y, Wang J, Lyu X, Yang R. Screening of a Bacillus subtilis strain producing both nattokinase and milk-clotting enzyme and its application in fermented milk with thrombolytic activity. J Dairy Sci. Jul 1 2021;doi:10.3168/jds.2020-19756
- Chandrasekaran SD, Vaithilingam M, Shanker R, et al. Exploring the In Vitro Thrombolytic Activity of Nattokinase From a New Strain Pseudomonas aeruginosa CMSS. Jundishapur journal of microbiology. Oct 2015;8(10):e23567. doi:10.5812/jjm.23567
- Hsia C-H, Shen M-C, Lin J-S, et al. Nattokinase decreases plasma levels of fibrinogen, factor VII, and factor VIII in human subjects. Nutrition Research. 2009;29(3):190-196.
- Cesarone MR, Belcaro G, Nicolaides AN, et al. Prevention of venous thrombosis in long-haul flights with Flite Tabs: the LONFLIT-FLITE randomized, controlled trial. Angiology. Sep-Oct 2003;54(5):531-9.
- Guo H, Ban YH, Cha Y, et al. Comparative anti-thrombotic activity and haemorrhagic adverse effect of nattokinase and tissue-type plasminogen activator. Food Sci Biotechnol. Oct 2019;28(5):1535-1542. doi:10.1007/s10068-019-00580-1
- Wu H, Wang Y, Zhang Y, et al. Breaking the vicious loop between inflammation, oxidative stress and coagulation, a novel anti-thrombus insight of nattokinase by inhibiting LPS-induced inflammation and oxidative stress. Redox biology. May 2020;32:101500. doi:10.1016/j.redox.2020.101500
- Jang JY, Kim TS, Cai J, et al. Nattokinase improves blood flow by inhibiting platelet aggregation and thrombus formation. Laboratory animal research. Dec 2013;29(4):221-5. doi:10.5625/lar.2013.29.4.221
- Kamiya S, Hagimori M, Ogasawara M, Arakawa M. In vivo evaluation method of the effect of nattokinase on carrageenan-induced tail thrombosis in a rat model. Acta haematologica. 2010;124(4):218-24. doi:10.1159/000321518
- Suzuki Y, Kondo K, Matsumoto Y, et al. Dietary supplementation of fermented soybean, natto, suppresses intimal thickening and modulates the lysis of mural thrombi after endothelial injury in rat femoral artery. Life Sci. Jul 25 2003;73(10):1289-98. doi:10.1016/s0024-3205(03)00426-0
- Suzuki Y, Kondo K, Ichise H, Tsukamoto Y, Urano T, Umemura K. Dietary supplementation with fermented soybeans suppresses intimal thickening. Nutrition (Burbank, Los Angeles County, Calif). Mar 2003;19(3):261-4. doi:10.1016/s0899-9007(02)00853-5
- Hodis HN, Mack WJ, Meiselman HJ, et al. Nattokinase atherothrombotic prevention study: A randomized controlled trial. Clinical hemorheology and microcirculation. Apr 2 2021;doi:10.3233/ch-211147
- Subedi BH, Joshi PH, Jones SR, Martin SS, Blaha MJ, Michos ED. Current guidelines for high-density lipoprotein cholesterol in therapy and future directions. Vasc Health Risk Manag. 2014;10:205-16. doi:10.2147/vhrm.S45648
- Ramirez A, Hu PP. Low High-Density Lipoprotein and Risk of Myocardial Infarction. Clinical Medicine Insights Cardiology. 2015;9:113-7. doi:10.4137/cmc.S26624
- Probstfield JL, Boden WE, Anderson T, et al. Cardiovascular outcomes during extended follow-up of the AIM-HIGH trial cohort. Journal of clinical lipidology. Nov-Dec 2018;12(6):1413-1419. doi:10.1016/j.jacl.2018.07.007
- Landray MJ, Haynes R, Hopewell JC, et al. Effects of extended-release niacin with laropiprant in high-risk patients. The New England journal of medicine. Jul 17 2014;371(3):203-12. doi:10.1056/NEJMoa1300955
- Zeman M, Vecka M, Perlík F, et al. Pleiotropic effects of niacin: Current possibilities for its clinical use. Acta pharmaceutica (Zagreb, Croatia). Dec 1 2016;66(4):449-469. doi:10.1515/acph-2016-0043
- Zhou K, Zhao R, Geng Z, et al. Association between B-group vitamins and venous thrombosis: systematic review and meta-analysis of epidemiological studies. Journal of thrombosis and thrombolysis. Nov 2012;34(4):459-67. doi:10.1007/s11239-012-0759-x
- Zaric BL, Obradovic M, Bajic V, Haidara MA, Jovanovic M, Isenovic ER. Homocysteine and Hyperhomocysteinaemia. Curr Med Chem. 2019;26(16):2948-2961. doi:10.2174/0929867325666180313105949
- Amaral FM, Miranda-Vilela AL, Lordelo GS, Ribeiro IF, Daldegan MB, Grisolia CK. Interactions among methylenetetrahydrofolate reductase (MTHFR) and cystathionine β-synthase (CBS) polymorphisms - a cross-sectional study: multiple heterozygosis as a risk factor for higher homocysteine levels and vaso-occlusive episodes. Genetics and molecular research : GMR. Feb 23 2017;16(1)doi:10.4238/gmr16019374
- Spence JD. Cardioembolic stroke: everything has changed. Stroke Vasc Neurol. Jun 2018;3(2):76-83. doi:10.1136/svn-2018-000143
- Ammouri W, Tazi ZM, Harmouche H, Maamar M, Adnaoui M. Venous thromboembolism and hyperhomocysteinemia as first manifestation of pernicious anemia: a case series. Journal of medical case reports. Sep 2 2017;11(1):250. doi:10.1186/s13256-017-1415-z
- Prajapati K, Sailor V, Patel S, Rathod M. Pernicious anaemia: cause of recurrent cerebral venous thrombosis. BMJ case reports. May 10 2021;14(5)doi:10.1136/bcr-2020-239833
- Spence JD. Homocysteine lowering for stroke prevention: Unravelling the complexity of the evidence. Int J Stroke. Oct 2016;11(7):744-7. doi:10.1177/1747493016662038
- Kong X, Huang X, Zhao M, et al. Platelet Count Affects Efficacy of Folic Acid in Preventing First Stroke. Journal of the American College of Cardiology. May 15 2018;71(19):2136-2146. doi:10.1016/j.jacc.2018.02.072
- Shu XJ, Li ZF, Chang YW, Liu SY, Wang WH. Effects of folic acid combined with vitamin B12 on DVT in patients with homocysteine cerebral infarction. European review for medical and pharmacological sciences. May 2017;21(10):2538-2544.
- Yamagata K. Protective Effect of Epigallocatechin Gallate on Endothelial Disorders in Atherosclerosis. Journal of cardiovascular pharmacology. Apr 2020;75(4):292-298. doi:10.1097/fjc.0000000000000792
- Slevin M, Ahmed N, Wang Q, McDowell G, Badimon L. Unique vascular protective properties of natural products: supplements or future main-line drugs with significant anti-atherosclerotic potential? Vasc Cell. Apr 30 2012;4(1):9. doi:10.1186/2045-824x-4-9
- Joo HJ, Park JY, Hong SJ, et al. Anti-platelet effects of epigallocatechin-3-gallate in addition to the concomitant aspirin, clopidogrel or ticagrelor treatment. The Korean journal of internal medicine. May 2018;33(3):522-531. doi:10.3904/kjim.2016.228
- Lee DH, Kim YJ, Kim HH, et al. Inhibitory effects of epigallocatechin-3-gallate on microsomal cyclooxygenase-1 activity in platelets. Biomolecules & therapeutics. Jan 2013;21(1):54-9. doi:10.4062/biomolther.2012.075
- Reddy AT, Lakshmi SP, Maruthi Prasad E, Varadacharyulu NC, Kodidhela LD. Epigallocatechin gallate suppresses inflammation in human coronary artery endothelial cells by inhibiting NF-κB. Life Sci. Oct 1 2020;258:118136. doi:10.1016/j.lfs.2020.118136
- Zhang Z, Zhang X, Bi K, et al. Potential protective mechanisms of green tea polyphenol EGCG against COVID-19. Trends Food Sci Technol. Aug 2021;114:11-24. doi:10.1016/j.tifs.2021.05.023
- İğde M, Onur Öztürk M, Yaşar B, Hakan Bulam M, Ergani HM, Ünlü RE. Antithrombotic effect of epigallocatechin gallate on the patency of arterial microvascular anastomoses. Archives of plastic surgery. May 2019;46(3):214-220. doi:10.5999/aps.2018.00157
- Lee DK, Grantham RN, Mannion JD, Trachte AL. Carotenoids enhance phosphorylation of Akt and suppress tissue factor activity in human endothelial cells. J Nutr Biochem. Nov 2006;17(11):780-6. doi:10.1016/j.jnutbio.2006.01.006
- Hsiao G, Wang Y, Tzu NH, et al. Inhibitory effects of lycopene on in vitro platelet activation and in vivo prevention of thrombus formation. The Journal of laboratory and clinical medicine. Oct 2005;146(4):216-26. doi:10.1016/j.lab.2005.03.018
- Sawardekar SB, Patel TC, Uchil D. Comparative evaluation of antiplatelet effect of lycopene with aspirin and the effect of their combination on platelet aggregation: An in vitro study. Indian journal of pharmacology. Jan-Feb 2016;48(1):26-31. doi:10.4103/0253-7613.174428
- Thies F, Mills LM, Moir S, Masson LF. Cardiovascular benefits of lycopene: fantasy or reality? The Proceedings of the Nutrition Society. May 2017;76(2):122-129. doi:10.1017/s0029665116000744
- Concha-Meyer A, Palomo I, Plaza A, et al. Platelet Anti-Aggregant Activity and Bioactive Compounds of Ultrasound-Assisted Extracts from Whole and Seedless Tomato Pomace. Foods. Oct 28 2020;9(11)doi:10.3390/foods9111564
- Fuentes E, Trostchansky A, Reguengo LM, Maróstica MR, Jr., Palomo I. Antiplatelet effects of bioactive compounds present in tomato pomace. Current drug targets. Jan 28 2021;doi:10.2174/1389450122999210128180456
- Palomo I, Concha-Meyer A, Lutz M, et al. Chemical Characterization and Antiplatelet Potential of Bioactive Extract from Tomato Pomace (Byproduct of Tomato Paste). Nutrients. Feb 22 2019;11(2)doi:10.3390/nu11020456
- Summerhill V, Karagodin V, Grechko A, Myasoedova V, Orekhov A. Vasculoprotective Role of Olive Oil Compounds via Modulation of Oxidative Stress in Atherosclerosis. Front Cardiovasc Med. 2018;5:188. doi:10.3389/fcvm.2018.00188
- Cicerale S, Lucas L, Keast R. Biological activities of phenolic compounds present in virgin olive oil. International journal of molecular sciences. Feb 2 2010;11(2):458-79. doi:10.3390/ijms11020458
- Peyrol J, Riva C, Amiot MJ. Hydroxytyrosol in the Prevention of the Metabolic Syndrome and Related Disorders. Nutrients. Mar 20 2017;9(3)doi:10.3390/nu9030306
- Mizutani D, Onuma T, Tanabe K, et al. Olive polyphenol reduces the collagen-elicited release of phosphorylated HSP27 from human platelets. Bioscience, biotechnology, and biochemistry. Mar 2020;84(3):536-543. doi:10.1080/09168451.2019.1697196
- Zbidi H, Salido S, Altarejos J, et al. Olive tree wood phenolic compounds with human platelet antiaggregant properties. Blood Cells Mol Dis. May-Jun 2009;42(3):279-85. doi:10.1016/j.bcmd.2009.01.001
- Dell'Agli M, Maschi O, Galli GV, et al. Inhibition of platelet aggregation by olive oil phenols via cAMP-phosphodiesterase. The British journal of nutrition. May 2008;99(5):945-51. doi:10.1017/s0007114507837470
- Singh I, Mok M, Christensen AM, Turner AH, Hawley JA. The effects of polyphenols in olive leaves on platelet function. Nutr Metab Cardiovasc Dis. Feb 2008;18(2):127-32. doi:10.1016/j.numecd.2006.09.001
- Fourati M, Smaoui S, Hlima HB, et al. Bioactive Compounds and Pharmacological Potential of Pomegranate (Punica granatum) Seeds - A Review. Plant foods for human nutrition (Dordrecht, Netherlands). Dec 2020;75(4):477-486. doi:10.1007/s11130-020-00863-7
- Chang Y, Chen WF, Lin KH, et al. Novel bioactivity of ellagic Acid in inhibiting human platelet activation. Evidence-based complementary and alternative medicine : eCAM. 2013;2013:595128. doi:10.1155/2013/595128
- Mattiello T, Trifirò E, Jotti GS, Pulcinelli FM. Effects of pomegranate juice and extract polyphenols on platelet function. Journal of medicinal food. Apr 2009;12(2):334-9. doi:10.1089/jmf.2007.0640
- Sahebkar A, Ferri C, Giorgini P, Bo S, Nachtigal P, Grassi D. Effects of pomegranate juice on blood pressure: A systematic review and meta-analysis of randomized controlled trials. Pharmacological research : the official journal of the Italian Pharmacological Society . Jan 2017;115:149-161. doi:10.1016/j.phrs.2016.11.018
- Aviram M, Dornfeld L, Rosenblat M, et al. Pomegranate juice consumption reduces oxidative stress, atherogenic modifications to LDL, and platelet aggregation: studies in humans and in atherosclerotic apolipoprotein E-deficient mice. Am J Clin Nutr. May 2000;71(5):1062-76. doi:10.1093/ajcn/71.5.1062
- Polagruto JA, Schramm DD, Wang-Polagruto JF, Lee L, Keen CL. Effects of flavonoid-rich beverages on prostacyclin synthesis in humans and human aortic endothelial cells: association with ex vivo platelet function. Journal of medicinal food. Winter 2003;6(4):301-8. doi:10.1089/109662003772519840
- Wang D, Ozen C, Abu-Reidah IM, et al. Vasculoprotective Effects of Pomegranate (Punica granatum L.). Front Pharmacol. 2018;9:544. doi:10.3389/fphar.2018.00544
- Delgado NTB, Rouver WN, Dos Santos RL. Protective Effects of Pomegranate in Endothelial Dysfunction. Curr Pharm Des. 2020;26(30):3684-3699. doi:10.2174/1381612826666200406152147
- Jafari T, Fallah AA, Reyhanian A, Sarmast E. Effects of pomegranate peel extract and vitamin E on the inflammatory status and endothelial function in hemodialysis patients: a randomized controlled clinical trial. Food Funct. Sep 23 2020;11(9):7987-7993. doi:10.1039/d0fo01012j
- Hosseini B, Saedisomeolia A, Wood LG, Yaseri M, Tavasoli S. Effects of pomegranate extract supplementation on inflammation in overweight and obese individuals: A randomized controlled clinical trial. Complementary therapies in clinical practice. Feb 2016;22:44-50. doi:10.1016/j.ctcp.2015.12.003
- Lutomski P, Goździewska M, Florek-Łuszczki M. Health properties of Yerba Mate. Ann Agric Environ Med. Jun 19 2020;27(2):310-313. doi:10.26444/aaem/119994
- Yu S, Yue S, Liu Z, Zhang T, Xiang N, Fu H. Yerba mate (Ilex paraguariensis) improves microcirculation of volunteers with high blood viscosity: a randomized, double-blind, placebo-controlled trial. Exp Gerontol. Feb 2015;62:14-22. doi:10.1016/j.exger.2014.12.016
- Gebara KS, Gasparotto Junior A, Palozi RAC, et al. A Randomized Crossover Intervention Study on the Effect a Standardized Maté Extract (Ilex paraguariensis A. St.-Hil.) in Men Predisposed to Cardiovascular Risk. Nutrients. Dec 23 2020;13(1)doi:10.3390/nu13010014
- Dahmer T, Berger M, Barlette AG, et al. Antithrombotic effect of chikusetsusaponin IVa isolated from Ilex paraguariensis (Maté). Journal of medicinal food. Dec 2012;15(12):1073-80. doi:10.1089/jmf.2011.0320
- Oranuba E, Deng H, Peng J, Dawsey SM, Kamangar F. Polycyclic aromatic hydrocarbons as a potential source of carcinogenicity of mate. Journal of environmental science and health Part C, Environmental carcinogenesis & ecotoxicology reviews . 2019;37(1):26-41. doi:10.1080/10590501.2019.1555323
- Anand David AV, Arulmoli R, Parasuraman S. Overviews of Biological Importance of Quercetin: A Bioactive Flavonoid. Pharmacogn Rev. Jul-Dec 2016;10(20):84-89. doi:10.4103/0973-7847.194044
- Hubbard GP, Wolffram S, Lovegrove JA, Gibbins JM. Ingestion of quercetin inhibits platelet aggregation and essential components of the collagen-stimulated platelet activation pathway in humans. J Thromb Haemost. Dec 2004;2(12):2138-45. doi:10.1111/j.1538-7836.2004.01067.x
- Egert S, Bosy-Westphal A, Seiberl J, et al. Quercetin reduces systolic blood pressure and plasma oxidised low-density lipoprotein concentrations in overweight subjects with a high-cardiovascular disease risk phenotype: a double-blinded, placebo-controlled cross-over study. The British journal of nutrition. Oct 2009;102(7):1065-74. doi:10.1017/s0007114509359127
- Patel RV, Mistry BM, Shinde SK, Syed R, Singh V, Shin HS. Therapeutic potential of quercetin as a cardiovascular agent. European journal of medicinal chemistry. Jul 15 2018;155:889-904. doi:10.1016/j.ejmech.2018.06.053
- Zhang YX, Yang TT, Xia L, Zhang WF, Wang JF, Wu YP. Inhibitory Effect of Propolis on Platelet Aggregation In Vitro. J Healthc Eng. 2017;2017:3050895. doi:10.1155/2017/3050895
- Olas B. Honey and Its Phenolic Compounds as an Effective Natural Medicine for Cardiovascular Diseases in Humans? Nutrients. Jan 21 2020;12(2)doi:10.3390/nu12020283
- Jessie SW, Krishnakantha TP. Inhibition of human platelet aggregation and membrane lipid peroxidation by food spice, saffron. Molecular and cellular biochemistry. Oct 2005;278(1-2):59-63. doi:10.1007/s11010-005-5155-9
- Su X, Yuan C, Wang L, et al. The Beneficial Effects of Saffron Extract on Potential Oxidative Stress in Cardiovascular Diseases. Oxid Med Cell Longev. 2021;2021:6699821. doi:10.1155/2021/6699821
- Tsantarliotou MP, Poutahidis T, Markala D, et al. Crocetin administration ameliorates endotoxin-induced disseminated intravascular coagulation in rabbits. Blood Coagul Fibrinolysis. Apr 2013;24(3):305-10. doi:10.1097/MBC.0b013e32835bdc8f
- Yang L, Qian Z, Yang Y, et al. Involvement of Ca2+ in the inhibition by crocetin of platelet activity and thrombosis formation. J Agric Food Chem. Oct 22 2008;56(20):9429-33. doi:10.1021/jf802027a
- Higashino S, Sasaki Y, Giddings JC, et al. Crocetin, a carotenoid from Gardenia jasminoides Ellis, protects against hypertension and cerebral thrombogenesis in stroke-prone spontaneously hypertensive rats. Phytother Res. Sep 2014;28(9):1315-9. doi:10.1002/ptr.5130
- Abedimanesh N, Motlagh B, Abedimanesh S, Bathaie SZ, Separham A, Ostadrahimi A. Effects of crocin and saffron aqueous extract on gene expression of SIRT1, AMPK, LOX1, NF-κB, and MCP-1 in patients with coronary artery disease: A randomized placebo-controlled clinical trial. Phytother Res. May 2020;34(5):1114-1122. doi:10.1002/ptr.6580
- Mao QQ, Xu XY, Cao SY, et al. Bioactive Compounds and Bioactivities of Ginger (Zingiber officinale Roscoe). Foods. May 30 2019;8(6)doi:10.3390/foods8060185
- Lee W, Ku SK, Kim MA, Bae JS. Anti-factor Xa activities of zingerone with anti-platelet aggregation activity. Food and chemical toxicology : an international journal published for the British Industrial Biological Research Association . Jul 2017;105:186-193. doi:10.1016/j.fct.2017.04.012
- Wang C, He Y, Tang X, Li N. Sulfation, structural analysis, and anticoagulant bioactivity of ginger polysaccharides. Journal of food science. Aug 2020;85(8):2427-2434. doi:10.1111/1750-3841.15338
- Bordia A, Verma SK, Srivastava KC. Effect of ginger (Zingiber officinale Rosc.) and fenugreek (Trigonella foenumgraecum L.) on blood lipids, blood sugar and platelet aggregation in patients with coronary artery disease. Prostaglandins Leukot Essent Fatty Acids. May 1997;56(5):379-84. doi:10.1016/s0952-3278(97)90587-1
- Liu D, Pei D, Hu H, Gu G, Cui W. Effects and Mechanisms of Vitamin C Post-Conditioning on Platelet Activation after Hypoxia/Reoxygenation. Transfus Med Hemother. Apr 2020;47(2):110-118. doi:10.1159/000500492
- Tyml K. Vitamin C and Microvascular Dysfunction in Systemic Inflammation. Antioxidants (Basel, Switzerland). Jun 29 2017;6(3)doi:10.3390/antiox6030049
- Mohammed BM, Sanford KW, Fisher BJ, et al. Impact of high dose vitamin C on platelet function. World journal of critical care medicine. Feb 4 2017;6(1):37-47. doi:10.5492/wjccm.v6.i1.37
- Secor D, Swarbreck S, Ellis CG, Sharpe MD, Feng Q, Tyml K. Ascorbate inhibits platelet-endothelial adhesion in an in-vitro model of sepsis via reduced endothelial surface P-selectin expression. Blood Coagul Fibrinolysis. Jan 2017;28(1):28-33. doi:10.1097/mbc.0000000000000528
- Ashor AW, Siervo M, Lara J, Oggioni C, Afshar S, Mathers JC. Effect of vitamin C and vitamin E supplementation on endothelial function: a systematic review and meta-analysis of randomised controlled trials. The British journal of nutrition. Apr 28 2015;113(8):1182-94. doi:10.1017/s0007114515000227
- Ashor AW, Brown R, Keenan PD, Willis ND, Siervo M, Mathers JC. Limited evidence for a beneficial effect of vitamin C supplementation on biomarkers of cardiovascular diseases: an umbrella review of systematic reviews and meta-analyses. Nutr Res. Jan 2019;61:1-12. doi:10.1016/j.nutres.2018.08.005
- Moser MA, Chun OK. Vitamin C and Heart Health: A Review Based on Findings from Epidemiologic Studies. International journal of molecular sciences. Aug 12 2016;17(8)doi:10.3390/ijms17081328
- Hornig B, Arakawa N, Kohler C, Drexler H. Vitamin C improves endothelial function of conduit arteries in patients with chronic heart failure. Circulation. Feb 3 1998;97(4):363-8. doi:10.1161/01.cir.97.4.363
- Reilly M, Delanty N, Lawson JA, FitzGerald GA. Modulation of oxidant stress in vivo in chronic cigarette smokers. Circulation. Jul 1 1996;94(1):19-25. doi:10.1161/01.cir.94.1.19
- Weber C, Erl W, Weber K, Weber PC. Increased adhesiveness of isolated monocytes to endothelium is prevented by vitamin C intake in smokers. Circulation. Apr 15 1996;93(8):1488-92. doi:10.1161/01.cir.93.8.1488
- Bijak M, Sut A, Kosiorek A, Saluk-Bijak J, Golanski J. Dual Anticoagulant/Antiplatelet Activity of Polyphenolic Grape Seeds Extract. Nutrients. Jan 5 2019;11(1)doi:10.3390/nu11010093
- Jin JW, Inoue O, Suzuki-Inoue K, et al. Grape seed extracts inhibit platelet aggregation by inhibiting protein tyrosine phosphatase. Clinical and applied thrombosis/hemostasis : official journal of the International Academy of Clinical and Applied Thrombosis/Hemostasis . Apr 2014;20(3):278-84. doi:10.1177/1076029613481103
- Zhang Y, Shi H, Wang W, et al. Antithrombotic effect of grape seed proanthocyanidins extract in a rat model of deep vein thrombosis. J Vasc Surg. Mar 2011;53(3):743-53. doi:10.1016/j.jvs.2010.09.017
- Zhang H, Liu S, Li L, et al. The impact of grape seed extract treatment on blood pressure changes: A meta-analysis of 16 randomized controlled trials. Medicine. Aug 2016;95(33):e4247. doi:10.1097/md.0000000000004247
- Sanati S, Razavi BM, Hosseinzadeh H. A review of the effects of Capsicum annuum L. and its constituent, capsaicin, in metabolic syndrome. Iranian journal of basic medical sciences. May 2018;21(5):439-448. doi:10.22038/ijbms.2018.25200.6238
- Panchal SK, Bliss E, Brown L. Capsaicin in Metabolic Syndrome. Nutrients. May 17 2018;10(5)doi:10.3390/nu10050630
- Fattori V, Hohmann MS, Rossaneis AC, Pinho-Ribeiro FA, Verri WA. Capsaicin: Current Understanding of Its Mechanisms and Therapy of Pain and Other Pre-Clinical and Clinical Uses. Molecules (Basel, Switzerland). Jun 28 2016;21(7)doi:10.3390/molecules21070844
- Almaghrabi S, Adams M, Geraghty D, Ahuja K. Synergistic inhibitory effect of capsaicin and dihydrocapsaicin on in-vitro platelet aggregation and thromboxane formation. Blood Coagul Fibrinolysis. Jun 2018;29(4):351-355. doi:10.1097/mbc.0000000000000698
- Wang JP, Hsu MF, Teng CM. Antiplatelet effect of capsaicin. Thromb Res. Dec 15 1984;36(6):497-507. doi:10.1016/0049-3848(84)90189-0
- Wang JP, Hsu MF, Hsu TP, Teng CM. Antihemostatic and antithrombotic effects of capsaicin in comparison with aspirin and indomethacin. Thromb Res. Mar 15 1985;37(6):669-79. doi:10.1016/0049-3848(85)90196-3
- Raghavendra RH, Naidu KA. Spice active principles as the inhibitors of human platelet aggregation and thromboxane biosynthesis. Prostaglandins Leukot Essent Fatty Acids. Jul 2009;81(1):73-8. doi:10.1016/j.plefa.2009.04.009
- Sandor B, Papp J, Mozsik G, et al. Orally given gastroprotective capsaicin does not modify aspirin-induced platelet aggregation in healthy male volunteers (human phase I examination). Acta physiologica Hungarica. Dec 2014;101(4):429-37. doi:10.1556/APhysiol.101.2014.4.4
- Breuss JM, Atanasov AG, Uhrin P. Resveratrol and Its Effects on the Vascular System. International journal of molecular sciences. Mar 27 2019;20(7)doi:10.3390/ijms20071523
- Snopek L, Mlcek J, Sochorova L, et al. Contribution of Red Wine Consumption to Human Health Protection. Molecules (Basel, Switzerland). Jul 11 2018;23(7)doi:10.3390/molecules23071684
- Wiciński M, Socha M, Walczak M, et al. Beneficial Effects of Resveratrol Administration-Focus on Potential Biochemical Mechanisms in Cardiovascular Conditions. Nutrients. Nov 21 2018;10(11)doi:10.3390/nu10111813
- Marumo M, Ekawa K, Wakabayashi I. Resveratrol inhibits Ca(2+) signals and aggregation of platelets. Environ Health Prev Med. Nov 7 2020;25(1):70. doi:10.1186/s12199-020-00905-1
- Xue Y, Chen H, Zhang S, et al. Resveratrol Confers Vascular Protection by Suppressing TLR4/Syk/NLRP3 Signaling in Oxidized Low-Density Lipoprotein-Activated Platelets. Oxid Med Cell Longev. 2021;2021:8819231. doi:10.1155/2021/8819231
- Sun J, Zhang M, Chen K, et al. Suppression of TLR4 activation by resveratrol is associated with STAT3 and Akt inhibition in oxidized low-density lipoprotein-activated platelets. European journal of pharmacology. Oct 5 2018;836:1-10. doi:10.1016/j.ejphar.2018.08.014
- Bonechi C, Lamponi S, Donati A, et al. Effect of resveratrol on platelet aggregation by fibrinogen protection. Biophysical chemistry. Mar 2017;222:41-48. doi:10.1016/j.bpc.2016.12.004
- Ratan ZA, Haidere MF, Hong YH, et al. Pharmacological potential of ginseng and its major component ginsenosides. J Ginseng Res. Mar 2021;45(2):199-210. doi:10.1016/j.jgr.2020.02.004
- Luo BY, Jiang JL, Fang YF, et al. The effects of ginsenosides on platelet aggregation and vascular intima in the treatment of cardiovascular diseases: From molecular mechanisms to clinical applications. Pharmacological research : the official journal of the Italian Pharmacological Society . Sep 2020;159:105031. doi:10.1016/j.phrs.2020.105031
- Wang MM, Xue M, Miao Y, et al. Panax quinquefolium saponin combined with dual antiplatelet drugs inhibits platelet adhesion to injured HUVECs via PI3K/AKT and COX pathways. Journal of ethnopharmacology. Nov 4 2016;192:10-19. doi:10.1016/j.jep.2016.07.015
- Wang J, Xu J, Zhong JB. [Effect of Radix notoginseng saponins on platelet activating molecule expression and aggregation in patients with blood hyperviscosity syndrome]. Zhongguo Zhong xi yi jie he za zhi Zhongguo Zhongxiyi jiehe zazhi = Chinese journal of integrated traditional and Western medicine / Zhongguo Zhong xi yi jie he xue hui, Zhongguo Zhong yi yan jiu yuan zhu ban . Apr 2004;24(4):312-6.
- Zuo X, Li Q, Ya F, et al. Ginsenosides Rb2 and Rd2 isolated from Panax notoginseng flowers attenuate platelet function through P2Y(12)-mediated cAMP/PKA and PI3K/Akt/Erk1/2 signaling. Food Funct. May 27 2021;doi:10.1039/d1fo00531f
- Xu ZY, Xu Y, Xie XF, et al. Anti-platelet aggregation of Panax notoginseng triol saponins by regulating GP1BA for ischemic stroke therapy. Chinese medicine. Jan 19 2021;16(1):12. doi:10.1186/s13020-021-00424-3
- Xue Q, He N, Wang Z, et al. Functional roles and mechanisms of ginsenosides from Panax ginseng in atherosclerosis. J Ginseng Res. Jan 2021;45(1):22-31. doi:10.1016/j.jgr.2020.07.002
- Yoon SJ, Kim SK, Lee NY, et al. Effect of Korean Red Ginseng on metabolic syndrome. J Ginseng Res. May 2021;45(3):380-389. doi:10.1016/j.jgr.2020.11.002
- Irfan M, Jeong D, Saba E, et al. Gintonin modulates platelet function and inhibits thrombus formation via impaired glycoprotein VI signaling. Platelets. 2019;30(5):589-598. doi:10.1080/09537104.2018.1479033
- Keihanian F, Saeidinia A, Bagheri RK, Johnston TP, Sahebkar A. Curcumin, hemostasis, thrombosis, and coagulation. J Cell Physiol. Jun 2018;233(6):4497-4511. doi:10.1002/jcp.26249
- Tabeshpour J, Hashemzaei M, Sahebkar A. The regulatory role of curcumin on platelet functions. Journal of cellular biochemistry. Nov 2018;119(11):8713-8722. doi:10.1002/jcb.27192
- Wang T, Guan R, Xia F, Du J, Xu L. Curcumin promotes venous thrombi resolve process in a mouse deep venous thrombosis model via regulating miR-499. Microvascular research. Jul 2021;136:104148. doi:10.1016/j.mvr.2021.104148
- Nemmar A, Subramaniyan D, Ali BH. Protective effect of curcumin on pulmonary and cardiovascular effects induced by repeated exposure to diesel exhaust particles in mice. PLoS One. 2012;7(6):e39554.
- Klafke JZ, Arnoldi da Silva M, Fortes Rossato M, et al. Antiplatelet, Antithrombotic, and Fibrinolytic Activities of Campomanesia xanthocarpa. Evidence-based complementary and alternative medicine : eCAM. 2012;2012:954748. doi:10.1155/2012/954748
- Cunha EBB, Silva NFD, Lima J, Serrato JA, Aita CAM, Herai RH. Leaf extracts of Campomanesia xanthocarpa positively regulates atherosclerotic-related protein expression. An Acad Bras Cienc. 2020;92(4):e20191486. doi:10.1590/0001-3765202020191486
- Otero JS, Hirsch GE, Klafke JZ, et al. Inhibitory effect of Campomanesia xanthocarpa in platelet aggregation: Comparison and synergism with acetylsalicylic acid. Thromb Res. Jun 2017;154:42-49. doi:10.1016/j.thromres.2017.03.020
- Viecili PR, Borges DO, Kirsten K, et al. Effects of Campomanesia xanthocarpa on inflammatory processes, oxidative stress, endothelial dysfunction and lipid biomarkers in hypercholesterolemic individuals. Atherosclerosis. May 2014;234(1):85-92. doi:10.1016/j.atherosclerosis.2014.02.010
- Klafke JZ, da Silva MA, Panigas TF, et al. Effects of Campomanesia xanthocarpa on biochemical, hematological and oxidative stress parameters in hypercholesterolemic patients. Journal of ethnopharmacology. Feb 3 2010;127(2):299-305. doi:10.1016/j.jep.2009.11.004
- Prandoni P. Venous and Arterial Thrombosis: Is There a Link? Adv Exp Med Biol. 2017;906:273-283. doi:10.1007/5584_2016_121
- Mi Y, Yan S, Lu Y, Liang Y, Li C. Venous thromboembolism has the same risk factors as atherosclerosis: A PRISMA-compliant systemic review and meta-analysis. Medicine. Aug 2016;95(32):e4495. doi:10.1097/md.0000000000004495
- Stevens H, McFadyen JD. Platelets as Central Actors in Thrombosis-Reprising an Old Role and Defining a New Character. Semin Thromb Hemost. Nov 2019;45(8):802-809. doi:10.1055/s-0039-1698829
- Kemp MT, Obi AT, Henke PK, Wakefield TW. A narrative review on the epidemiology, prevention, and treatment of venous thromboembolic events in the context of chronic venous disease. Journal of vascular surgery Venous and lymphatic disorders. Apr 16 2021;doi:10.1016/j.jvsv.2021.03.018
- Matic M, Matic A, Djuran V, Gajinov Z, Prcic S, Golusin Z. Frequency of Peripheral Arterial Disease in Patients With Chronic Venous Insufficiency. Iran Red Crescent Med J. Jan 2016;18(1):e20781. doi:10.5812/ircmj.20781
- Khatana C, Saini NK, Chakrabarti S, et al. Mechanistic Insights into the Oxidized Low-Density Lipoprotein-Induced Atherosclerosis. Oxid Med Cell Longev. 2020;2020:5245308. doi:10.1155/2020/5245308
- Pathak RK, Abhilash SP, Hendriks JM. A Team-Based Approach Toward Risk Factors of Atrial Fibrillation. Card Electrophysiol Clin. Mar 2021;13(1):257-262. doi:10.1016/j.ccep.2020.11.008
- Hu YF, Chen YJ, Lin YJ, Chen SA. Inflammation and the pathogenesis of atrial fibrillation. Nature reviews Cardiology. Apr 2015;12(4):230-43. doi:10.1038/nrcardio.2015.2
- Habib A, Petrucci G, Rocca B. Pathophysiology of Thrombosis in Peripheral Artery Disease. Curr Vasc Pharmacol. 2020;18(3):204-214. doi:10.2174/1570161117666190206234046
- Ammermann F, Meinel FG, Beller E, et al. Concomitant chronic venous insufficiency in patients with peripheral artery disease: insights from MR angiography. European radiology. Jul 2020;30(7):3908-3914. doi:10.1007/s00330-020-06696-x
- Gutmann C, Siow R, Gwozdz AM, Saha P, Smith A. Reactive Oxygen Species in Venous Thrombosis. International journal of molecular sciences. Mar 11 2020;21(6)doi:10.3390/ijms21061918
- Obermayer G, Afonyushkin T, Binder CJ. Oxidized low-density lipoprotein in inflammation-driven thrombosis. J Thromb Haemost. Mar 2018;16(3):418-428. doi:10.1111/jth.13925
- Sloop GD, Weidman JJ, St Cyr JA. Atherothrombosis is a Thrombotic, not Inflammatory Disease. Cureus. Dec 5 2017;9(12):e1909. doi:10.7759/cureus.1909
- Pechlivani N, Ajjan RA. Thrombosis and Vascular Inflammation in Diabetes: Mechanisms and Potential Therapeutic Targets. Front Cardiovasc Med . 2018;5:1. doi:10.3389/fcvm.2018.00001
- Santilli F, Davì G, Patrono C. Homocysteine, methylenetetrahydrofolate reductase, folate status and atherothrombosis: A mechanistic and clinical perspective. Vascul Pharmacol. Mar 2016;78:1-9. doi:10.1016/j.vph.2015.06.009
- Yao Y, Shang MS, Dong JZ, Ma CS. Homocysteine in non-valvular atrial fibrillation: Role and clinical implications. Clin Chim Acta. Dec 2017;475:85-90. doi:10.1016/j.cca.2017.10.012
- Bikov A, Meszaros M, Schwarz EI. Coagulation and Fibrinolysis in Obstructive Sleep Apnoea. International journal of molecular sciences. Mar 11 2021;22(6)doi:10.3390/ijms22062834
- Man AWC, Li H, Xia N. Circadian Rhythm: Potential Therapeutic Target for Atherosclerosis and Thrombosis. International journal of molecular sciences. Jan 12 2021;22(2)doi:10.3390/ijms22020676
- De Stefano V. Arterial thrombosis and cancer: the neglected side of the coin of Trousseau syndrome. Haematologica. Sep 2018;103(9):1419-1421. doi:10.3324/haematol.2018.197814
- Obermann WMJ, Brockhaus K, Eble JA. Platelets, Constant and Cooperative Companions of Sessile and Disseminating Tumor Cells, Crucially Contribute to the Tumor Microenvironment. Frontiers in cell and developmental biology. 2021;9:674553. doi:10.3389/fcell.2021.674553
- Cumbler E. In-Hospital Ischemic Stroke. Neurohospitalist. Jul 2015;5(3):173-81. doi:10.1177/1941874415588319
- Lippi G, Favaloro EJ. Car Travel-Related Thrombosis: Fact or Fiction? Semin Thromb Hemost. Jun 2018;44(4):327-333. doi:10.1055/s-0038-1654716
- Cannegieter SC. Travel-related thrombosis. Best Pract Res Clin Haematol. Sep 2012;25(3):345-50. doi:10.1016/j.beha.2012.07.008
- Lippi G, Mattiuzzi C, Favaloro EJ. e-thrombosis: epidemiology, physiopathology and rationale for preventing computer-related thrombosis. Ann Transl Med. Sep 2018;6(17):344. doi:10.21037/atm.2018.09.03
- Rambaran KA, Alzghari SK. Gamer's Thrombosis: A Review of Published Reports. Ochsner J. Summer 2020;20(2):182-186. doi:10.31486/toj.19.0058
- Elbers LPB, Squizzato A, Gerdes VEA. Thyroid Disorders and Hemostasis. Semin Thromb Hemost. Oct 2018;44(7):676-682. doi:10.1055/s-0038-1666825
- Elbers LPB, Fliers E, Cannegieter SC. The influence of thyroid function on the coagulation system and its clinical consequences. J Thromb Haemost. Apr 2018;16(4):634-645. doi:10.1111/jth.13970
- Martinez JA, Qeadan F, Burge MR. Hypothyroidism, Sex, and Age Predict Future Thromboembolic Events Among Younger People. J Clin Endocrinol Metab. Apr 1 2020;105(4):e1593-600. doi:10.1210/clinem/dgz291
- Wei WT, Liu PP, Lin SM, et al. Hypothyroidism and the Risk of Venous Thromboembolism: A Nationwide Cohort Study. Thrombosis and haemostasis. Mar 2020;120(3):505-514. doi:10.1055/s-0039-3402761
- Oliver-Williams C, Glisic M, Shahzad S, et al. The route of administration, timing, duration and dose of postmenopausal hormone therapy and cardiovascular outcomes in women: a systematic review. Human reproduction update. Mar 1 2019;25(2):257-271. doi:10.1093/humupd/dmy039
- Gynecologists; TACoOa. Postmenopausal Estrogen Therapy Route of Administration and Risk of Venous Thromboembolism. Accessed September 9, 2021, https://www.acog.org/clinical/clinical-guidance/committee-opinion/articles/2013/04/postmenopausal-estrogen-therapy-route-of-administration-and-risk-of-venous-thromboembolism
- Marchetti M, Zermatten MG, Bertaggia Calderara D, Aliotta A, Alberio L. Heparin-Induced Thrombocytopenia: A Review of New Concepts in Pathogenesis, Diagnosis, and Management. J Clin Med. Feb 10 2021;10(4)doi:10.3390/jcm10040683
- Novakovic M, Rout A, Kingsley T, et al. Role of gut microbiota in cardiovascular diseases. World J Cardiol. Apr 26 2020;12(4):110-122. doi:10.4330/wjc.v12.i4.110
- Jansen VL, Gerdes VE, Middeldorp S, van Mens TE. Gut microbiota and their metabolites in cardiovascular disease. Best practice & research Clinical endocrinology & metabolism . Feb 10 2021:101492. doi:10.1016/j.beem.2021.101492
- Reinhardt C. The Gut Microbiota as an Influencing Factor of Arterial Thrombosis. Hamostaseologie. Jun 2019;39(2):173-179. doi:10.1055/s-0038-1675357
- Hasan RA, Koh AY, Zia A. The gut microbiome and thromboembolism. Thromb Res. May 2020;189:77-87. doi:10.1016/j.thromres.2020.03.003
- Pignatelli P, Fabietti G, Ricci A, Piattelli A, Curia MC. How Periodontal Disease and Presence of Nitric Oxide Reducing Oral Bacteria Can Affect Blood Pressure. International journal of molecular sciences. Oct 13 2020;21(20)doi:10.3390/ijms21207538
- Aydin S. Can vitamin K synthesis altered by dysbiosis of microbiota be blamed in the etiopathogenesis of venous thrombosis? Bioscience of microbiota, food and health. 2017;36(3):73-74. doi:10.12938/bmfh.17-007
- Camelo-Castillo A, Rivera-Caravaca JM, Orenes-Piñero E, et al. Gut Microbiota and the Quality of Oral Anticoagulation in Vitamin K Antagonists Users: A Review of Potential Implications. J Clin Med. Feb 11 2021;10(4)doi:10.3390/jcm10040715
- Baker M, Anjum F, dela Cruz J. Deep Venous Thrombosis Ultrasound Evaluation. StatPearls. StatPearls Publishing Copyright © 2021, StatPearls Publishing LLC.; 2021.
- DeVon HA, Mirzaei S, Zègre‐Hemsey J. Typical and Atypical Symptoms of Acute Coronary Syndrome: Time to Retire the Terms? Journal of the American Heart Association. 2020;9(7):e015539. doi:doi:10.1161/JAHA.119.015539
- Cehofski LJ, Honoré B, Vorum H. A Review: Proteomics in Retinal Artery Occlusion, Retinal Vein Occlusion, Diabetic Retinopathy and Acquired Macular Disorders. International journal of molecular sciences. Apr 28 2017;18(5)doi:10.3390/ijms18050907
- Wang KL, Chu PH, Lee CH, et al. Management of Venous Thromboembolisms: Part I. The Consensus for Deep Vein Thrombosis. Acta Cardiol Sin. Jan 2016;32(1):1-22. doi:10.6515/acs20151228a
- Anghel L, Sascău R, Radu R, Stătescu C. From Classical Laboratory Parameters to Novel Biomarkers for the Diagnosis of Venous Thrombosis. International journal of molecular sciences. Mar 11 2020;21(6)doi:10.3390/ijms21061920
- Yang X, Zhang D, Zhao Y, et al. Association between serum level of C-reactive protein and risk of cardiovascular events based on cohort studies. Journal of human hypertension. May 12 2021;doi:10.1038/s41371-021-00546-z
- Sethwala AM, Goh I, Amerena JV. Combating Inflammation in Cardiovascular Disease. Heart Lung Circ. Feb 2021;30(2):197-206. doi:10.1016/j.hlc.2020.09.003
- Hulshof AM, Hemker HC, Spronk HMH, Henskens YMC, Ten Cate H. Thrombin-Fibrin(ogen) Interactions, Host Defense and Risk of Thrombosis. International journal of molecular sciences. Mar 4 2021;22(5)doi:10.3390/ijms22052590
- Simes J, Robledo KP, White HD, et al. D-Dimer Predicts Long-Term Cause-Specific Mortality, Cardiovascular Events, and Cancer in Patients With Stable Coronary Heart Disease. Circulation. 2018;138(7):712-723. doi:doi:10.1161/CIRCULATIONAHA.117.029901
- Ohara T, Farhoudi M, Bang OY, Koga M, Demchuk AM. The emerging value of serum D-dimer measurement in the work-up and management of ischemic stroke. Int J Stroke. Feb 2020;15(2):122-131. doi:10.1177/1747493019876538
- Harris WS, Del Gobbo L, Tintle NL. The Omega-3 Index and relative risk for coronary heart disease mortality: Estimation from 10 cohort studies. Atherosclerosis. Jul 2017;262:51-54. doi:10.1016/j.atherosclerosis.2017.05.007
- Cismaru G, Serban T, Tirpe A. Ultrasound Methods in the Evaluation of Atherosclerosis: From Pathophysiology to Clinic. Biomedicines. Apr 13 2021;9(4)doi:10.3390/biomedicines9040418
- Cademartiri F, Balestrieri A, Cau R, et al. Insight from imaging on plaque vulnerability: similarities and differences between coronary and carotid arteries-implications for systemic therapies. Cardiovascular diagnosis and therapy. Aug 2020;10(4):1150-1162. doi:10.21037/cdt-20-528
- Kearon C, Kahn SR. Long-term treatment of venous thromboembolism. Blood. Jan 30 2020;135(5):317-325. doi:10.1182/blood.2019002364
- Baig MU, Bodle J. Thrombolytic Therapy. StatPearls. StatPearls Publishing Copyright © 2021, StatPearls Publishing LLC.; 2021.
- Li X, Du H, Song Z, et al. Efficacy and safety of sonothrombolysis in patients with acute ischemic stroke: A systematic review and meta-analysis. J Neurol Sci. Sep 15 2020;416:116998. doi:10.1016/j.jns.2020.116998
- Goel L, Jiang X. Advances in Sonothrombolysis Techniques Using Piezoelectric Transducers. Sensors (Basel, Switzerland). Feb 27 2020;20(5)doi:10.3390/s20051288
- Signorelli SS, Platania I, Tomasello SD, et al. Insights from Experiences on Antiplatelet Drugs in Stroke Prevention: A Review. International journal of environmental research and public health. Aug 12 2020;17(16)doi:10.3390/ijerph17165840
- Cho SW, Franchi F, Angiolillo DJ. Role of oral anticoagulant therapy for secondary prevention in patients with stable atherothrombotic disease manifestations. Therapeutic advances in hematology. 2019;10:2040620719861475. doi:10.1177/2040620719861475
- Raber I, McCarthy CP, Vaduganathan M, et al. The rise and fall of aspirin in the primary prevention of cardiovascular disease. Lancet. May 25 2019;393(10186):2155-2167. doi:10.1016/s0140-6736(19)30541-0
- Hackam DG, Spence JD. Antiplatelet Therapy in Ischemic Stroke and Transient Ischemic Attack. Stroke. 2019;50(3):773-778. doi:doi:10.1161/STROKEAHA.118.023954
- Kumano O, Akatsuchi K, Amiral J. Updates on Anticoagulation and Laboratory Tools for Therapy Monitoring of Heparin, Vitamin K Antagonists and Direct Oral Anticoagulants. Biomedicines. Mar 7 2021;9(3)doi:10.3390/biomedicines9030264
- Hong J, Ahn SY, Lee YJ, et al. Updated recommendations for the treatment of venous thromboembolism. Blood Res. Mar 31 2021;56(1):6-16. doi:10.5045/br.2021.2020083
- Batta A, Kalra BS, Khirasaria R. Critical Issues and Recent Advances in Anticoagulant Therapy: A Review. Neurology India. Sep-Oct 2019;67(5):1200-1212. doi:10.4103/0028-3886.271256
- Hunt BJ, Levi M. Urgent reversal of vitamin K antagonists. BMJ (Clinical research ed). 2018;360:j5424. doi:10.1136/bmj.j5424
- Schwalfenberg GK. Vitamins K1 and K2: The Emerging Group of Vitamins Required for Human Health. J Nutr Metab. 2017;2017:6254836. doi:10.1155/2017/6254836
- Shioi A, Morioka T, Shoji T, Emoto M. The Inhibitory Roles of Vitamin K in Progression of Vascular Calcification. Nutrients. Feb 23 2020;12(2)doi:10.3390/nu12020583
- Boonyawat K, Wang L, Lazo-Langner A, et al. The effect of low-dose oral vitamin K supplementation on INR stability in patients receiving warfarin. A randomised trial. Thrombosis and haemostasis. Aug 30 2016;116(3):480-5. doi:10.1160/th16-04-0320
- Majeed H, Rodger M, Forgie M, et al. Effect of 200μG/day of vitamin K1 on the variability of anticoagulation control in patients on warfarin: a randomized controlled trial. Thromb Res. Sep 2013;132(3):329-35. doi:10.1016/j.thromres.2013.07.019
- Sconce E, Avery P, Wynne H, Kamali F. Vitamin K supplementation can improve stability of anticoagulation for patients with unexplained variability in response to warfarin. Blood. Mar 15 2007;109(6):2419-23. doi:10.1182/blood-2006-09-049262
- Lam J, Schulman S, Witt DM, Vandvik PO, Qayyum F, Holbrook AM. Anticoagulation control with daily low-dose vitamin k to reduce clinically adverse outcomes and international normalized ratio variability: a systematic review and meta-analysis. Pharmacotherapy. Nov 2013;33(11):1184-90. doi:10.1002/phar.1302
- Levy DS, Grewal R, Le TH. Vitamin K deficiency: an emerging player in the pathogenesis of vascular calcification and an iatrogenic consequence of therapies in advanced renal disease. American journal of physiology Renal physiology. Oct 1 2020;319(4):F618-f623. doi:10.1152/ajprenal.00278.2020
- Chen A, Stecker E, B AW. Direct Oral Anticoagulant Use: A Practical Guide to Common Clinical Challenges. J Am Heart Assoc. Jul 7 2020;9(13):e017559. doi:10.1161/jaha.120.017559
- Machin M, Salim S, Tan M, Onida S, Davies AH, Shalhoub J. Surgical and non-surgical approaches in the management of lower limb post-thrombotic syndrome. Expert review of cardiovascular therapy. Mar 2021;19(3):191-200. doi:10.1080/14779072.2021.1876563
- da Silva LF, Porto MSR, de Sousa AB, Avena KM. Graduated compression stockings as a prophylactic measure in venous thromboembolism and edema of lower limbs triggered by air travel: a systematic review of clinical trials. J Vasc Bras. May 10 2021;20:e20200164. doi:10.1590/1677-5449.200164
- Galanaud JP, Genty-Vermorel C, Rolland C, et al. Compression stockings to prevent postthrombotic syndrome: Literature overview and presentation of the CELEST trial. Research and practice in thrombosis and haemostasis. Nov 2020;4(8):1239-1250. doi:10.1002/rth2.12445
- Tripodi A, Braham S, Scimeca B, Moia M, Peyvandi F. How and when to measure anticoagulant effects of direct oral anticoagulants? Practical issues. Pol Arch Intern Med. Jun 29 2018;128(6):379-385. doi:10.20452/pamw.4287
- Orme R, Judge HM, Storey RF. Monitoring Antiplatelet Therapy. Semin Thromb Hemost. Apr 2017;43(3):311-319. doi:10.1055/s-0036-1597298
- Shao T, Cheng Y, Jin J, et al. A comparison of three platelet function tests in ischemic stroke patients with antiplatelet therapy. Journal of clinical neuroscience : official journal of the Neurosurgical Society of Australasia . Aug 2020;78:91-96. doi:10.1016/j.jocn.2020.06.004
- Alvitigala BY, Gooneratne LV, Constantine GR, Wijesinghe R, Arawwawala L. Pharmacokinetic, pharmacodynamic, and pharmacogenetic assays to monitor clopidogrel therapy. Pharmacology research & perspectives. Dec 2020;8(6):e00686. doi:10.1002/prp2.686
- Bader KB, Bouchoux G, Holland CK. Sonothrombolysis. Adv Exp Med Biol. 2016;880:339-62. doi:10.1007/978-3-319-22536-4_19
- Lorenzatti A, Servato ML. Role of Anti-inflammatory Interventions in Coronary Artery Disease: Understanding the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). European cardiology. Aug 2018;13(1):38-41. doi:10.15420/ecr.2018.11.1
- Libby P. Targeting Inflammatory Pathways in Cardiovascular Disease: The Inflammasome, Interleukin-1, Interleukin-6 and Beyond. Cells. Apr 20 2021;10(4)doi:10.3390/cells10040951
- Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. The New England journal of medicine. Sep 21 2017;377(12):1119-1131. doi:10.1056/NEJMoa1707914
- Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet. Jan 27 2018;391(10118):319-328. doi:10.1016/s0140-6736(17)32814-3
- Libby P, Glynn R, Thuren T, Hilkert R, Ridker P. 358Understanding and mitigating the risk of infection with canakinumab. European Heart Journal. 2018;39(suppl_1)doi:10.1093/eurheartj/ehy564.358
- Verhamme P, Yi BA, Segers A, et al. Abelacimab for Prevention of Venous Thromboembolism. The New England journal of medicine. Aug 12 2021;385(7):609-617. doi:10.1056/NEJMoa2105872
- USNLM. US National Library of Medicine: Clinical Trials.gov: Safety and Tolerability of Abelacimab (MAA868) vs. Rivaroxaban in Patients With Atrial Fibrillation (AZALEA-TIMI 71). Available at https://clinicaltrials.gov/ct2/show/NCT04755283 . Last updated 08/11/2021. Accessed 08/18/2021. . 2021;
- Collins R, Reith C, Emberson J, et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet. Nov 19 2016;388(10059):2532-2561. doi:10.1016/s0140-6736(16)31357-5
- Navarese EP, Robinson JG, Kowalewski M, et al. Association Between Baseline LDL-C Level and Total and Cardiovascular Mortality After LDL-C Lowering: A Systematic Review and Meta-analysis. Jama. Apr 17 2018;319(15):1566-1579. doi:10.1001/jama.2018.2525
- Joseph P, Glynn R, Lonn E, et al. Rosuvastatin for the prevention of venous thromboembolism: a pooled analysis of the HOPE-3 and JUPITER randomized controlled trials. Cardiovasc Res. Mar 10 2021;doi:10.1093/cvr/cvab078
- Wallace A, Albadawi H, Hoang P, et al. Statins as a preventative therapy for venous thromboembolism. Cardiovascular diagnosis and therapy. Dec 2017;7(Suppl 3):S207-s218. doi:10.21037/cdt.2017.09.12
- Li R, Yuan M, Yu S, et al. Effect of statins on the risk of recurrent venous thromboembolism: A systematic review and meta-analysis. Pharmacological research : the official journal of the Italian Pharmacological Society . Mar 2021;165:105413. doi:10.1016/j.phrs.2020.105413
- Kronenberg RM, Beglinger S, Stalder O, et al. Statin therapy and recurrent venous thromboembolism in the elderly: a prospective cohort study. Sci Rep. Oct 15 2019;9(1):14804. doi:10.1038/s41598-019-51374-8
- Makedonov I, Kahn SR, Galanaud JP. Prevention and Management of the Post-Thrombotic Syndrome. J Clin Med. Mar 27 2020;9(4)doi:10.3390/jcm9040923
- Sahebkar A, Catena C, Ray KK, et al. Impact of statin therapy on plasma levels of plasminogen activator inhibitor-1. A systematic review and meta-analysis of randomised controlled trials. Thrombosis and haemostasis. Jul 4 2016;116(1):162-71. doi:10.1160/th15-10-0770
- Ferrari F, Martins VM, Teixeira M, Santos RD, Stein R. COVID-19 and Thromboinflammation: Is There a Role for Statins? Clinics (Sao Paulo, Brazil). 2021;76:e2518. doi:10.6061/clinics/2021/e2518
- Pawlos A, Niedzielski M, Gorzelak-Pabiś P, Broncel M, Woźniak E. COVID-19: Direct and Indirect Mechanisms of Statins. International journal of molecular sciences. Apr 17 2021;22(8)doi:10.3390/ijms22084177
- Ramkumar S, Raghunath A, Raghunath S. Statin Therapy: Review of Safety and Potential Side Effects. Acta Cardiol Sin. Nov 2016;32(6):631-639. doi:10.6515/acs20160611a
- Ma J, Chen X. Anti-inflammatory Therapy for Coronary Atherosclerotic Heart Disease: Unanswered Questions Behind Existing Successes. Front Cardiovasc Med. 2020;7:631398. doi:10.3389/fcvm.2020.631398
- Nidorf SM, Fiolet ATL, Mosterd A, et al. Colchicine in Patients with Chronic Coronary Disease. The New England journal of medicine. Nov 5 2020;383(19):1838-1847. doi:10.1056/NEJMoa2021372
- Tardif JC, Kouz S, Waters DD, et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. The New England journal of medicine. Dec 26 2019;381(26):2497-2505. doi:10.1056/NEJMoa1912388
- Xia M, Yang X, Qian C. Meta-analysis Evaluating the Utility of Colchicine in Secondary Prevention of Coronary Artery Disease. The American journal of cardiology. Feb 1 2021;140:33-38. doi:10.1016/j.amjcard.2020.10.043
- Masson W, Lobo M, Molinero G, Masson G, Lavalle-Cobo A. Role of Colchicine in Stroke Prevention: An Updated Meta-Analysis. J Stroke Cerebrovasc Dis. May 2020;29(5):104756. doi:10.1016/j.jstrokecerebrovasdis.2020.104756
- Cimmino G, Conte S, Morello A, et al. Colchicine inhibits the prothrombotic effects of oxLDL in human endothelial cells. Vascul Pharmacol. Apr 2021;137:106822. doi:10.1016/j.vph.2020.106822
- Malik J, Javed N, Ishaq U, Khan U, Laique T. Is There a Role for Colchicine in Acute Coronary Syndromes? A Literature Review. Cureus. May 17 2020;12(5):e8166. doi:10.7759/cureus.8166
- Spartalis M, Spartalis E, Tzatzaki E, et al. The Beneficial Therapy with Colchicine for Atherosclerosis via Anti-inflammation and Decrease in Hypertriglyceridemia. Cardiovascular & hematological agents in medicinal chemistry. 2018;16(2):74-80. doi:10.2174/1871525717666181211110332
- Lu DY, Huang CC, Huang PH, et al. Metformin use in patients with type 2 diabetes mellitus is associated with reduced risk of deep vein thrombosis: a non-randomized, pair-matched cohort study. BMC cardiovascular disorders. Dec 15 2014;14:187. doi:10.1186/1471-2261-14-187
- Wilson JM, Farley KX, Broida SE, Bradbury TL, Guild GN. Metformin Use Is Associated with Fewer Complications in Patients with Type-2 Diabetes Undergoing Total Knee Arthroplasty: A Propensity Score-Matched Analysis. The Journal of bone and joint surgery American volume. Apr 7 2021;103(7):601-608. doi:10.2106/jbjs.20.01535
- Witkowski M, Friebel J, Tabaraie T, et al. Metformin Is Associated with Reduced Tissue Factor Procoagulant Activity in Patients with Poorly Controlled Diabetes. Cardiovasc Drugs Ther. Sep 17 2020;doi:10.1007/s10557-020-07040-7
- Markowicz-Piasecka M, Sadkowska A, Huttunen KM, Podsiedlik M, Mikiciuk-Olasik E, Sikora J. An investigation into the pleiotropic activity of metformin. A glimpse of haemostasis. European journal of pharmacology. Apr 5 2020;872:172984. doi:10.1016/j.ejphar.2020.172984
- Annamaraju P, Baradhi KM. Pentoxifylline. StatPearls. StatPearls Publishing Copyright © 2021, StatPearls Publishing LLC.; 2021.
- Janaki S. Pentoxifylline in strokes: a clinical study. The Journal of international medical research. 1980;8(1):56-62. doi:10.1177/030006058000800110
- Lucas MA. Prevention of post-operative thrombosis in peripheral arteriopathies. Pentoxifylline vs. conventional antiaggregants: a six-month randomized follow-up study. Angiology. Jul 1984;35(7):443-50. doi:10.1177/000331978403500707
- Radmilović A, Borić Z, Naumović T, Stamenković M, Muśikić P. Shunt thrombosis prevention in hemodialysis patients--a double-blind, randomized study: pentoxifylline vs placebo. Angiology. Jul 1987;38(7):499-506. doi:10.1177/000331978703800701
- Adel M, Awad HA, Abdel-Naim AB, Al-Azizi MM. Effects of pentoxifylline on coagulation profile and disseminated intravascular coagulation incidence in Egyptian septic neonates. J Clin Pharm Ther. Jun 2010;35(3):257-65. doi:10.1111/j.1365-2710.2009.01077.x
- Ozden MGN, Koksal G, Oz H. Comparison of Antithrombin III and Pentoxifylline Treatments in Gram Negative Sepsis Patients Developing Disseminated Intravascular Coagulation. Medeni Med J. 2019;34(3):233-238. doi:10.5222/mmj.2019.05935
- Moriau M, Lavenne-Pardonge E, Crasborn L, von Frenckell R, Col-Debeys C. The treatment of severe or recurrent deep venous thrombosis. Beneficial effect of the co-administration of antiplatelet agents with or without rheological effects, and anticoagulants. Thromb Res. Jun 15 1995;78(6):469-82. doi:10.1016/0049-3848(95)00081-2
- Zhang X, Wu J, Zhang B. Xuesaitong injection as one adjuvant treatment of acute cerebral infarction: a systematic review and meta-analysis. BMC Complement Altern Med. Feb 27 2015;15:36. doi:10.1186/s12906-015-0560-4
- Yan ST, Gao F, Dong TW, et al. Meta-Analysis of Randomized Controlled Trials of Xueshuantong Injection in Prevention of Deep Venous Thrombosis of Lower Extremity after Orthopedic Surgery. Evidence-based complementary and alternative medicine : eCAM. 2020;2020:8877791. doi:10.1155/2020/8877791
- Li W, Xu F, Huang R, et al. Xueshuantong Injection in Treating Deep Venous Thrombosis: A Systematic Review and Trial Sequential Analysis. Evidence-based complementary and alternative medicine : eCAM. 2021;2021:6622925. doi:10.1155/2021/6622925
- Nasiri A, AlQahtani A, Rayes NH, AlQahtani R, Alkharras R, Alghethber H. Direct oral anticoagulant: Review article. Journal of family medicine and primary care . Aug 2022;11(8):4180-4183. doi:10.4103/jfmpc.jfmpc_2253_21. https://www.ncbi.nlm.nih.gov/pubmed/36352947
- Navar AM, Kolkailah AA, Overton R, et al. Trends in Oral Anticoagulant Use Among 436 864 Patients With Atrial Fibrillation in Community Practice, 2011 to 2020. J Am Heart Assoc. Nov 15 2022;11(22):e026723. doi:10.1161/JAHA.122.026723. https://www.ncbi.nlm.nih.gov/pubmed/36346063
- Paravattil B, Elewa H. Approaches to Direct Oral Anticoagulant Selection in Practice. J Cardiovasc Pharmacol Ther. Aug 9 2018:1074248418793137. doi:10.1177/1074248418793137. https://www.ncbi.nlm.nih.gov/pubmed/30092658
- Grymonprez M, De Backer TL, Bertels X, Steurbaut S, Lahousse L. Long-term comparative effectiveness and safety of dabigatran, rivaroxaban, apixaban and edoxaban in patients with atrial fibrillation: A nationwide cohort study. Frontiers in pharmacology. 2023;14:1125576. doi:10.3389/fphar.2023.1125576. https://www.ncbi.nlm.nih.gov/pubmed/36817122
- Rogula S, Gasecka A, Mazurek T, Navarese EP, Szarpak L, Filipiak KJ. Safety and Efficacy of DOACs in Patients with Advanced and End-Stage Renal Disease. International journal of environmental research and public health . Jan 27 2022;19(3)doi:10.3390/ijerph19031436. https://www.ncbi.nlm.nih.gov/pubmed/35162472
- Mainbourg S, Cucherat M, Provencher S, et al. Twice- or Once-Daily Dosing of Direct Oral Anticoagulants, a systematic review and meta-analysis. Thromb Res. Jan 2021;197:24-32. doi:10.1016/j.thromres.2020.10.011. https://www.ncbi.nlm.nih.gov/pubmed/33161284
- Glise Sandblad K, Schulman S, Rosengren A, Sorbo J, Philipson J, Hansson PO. Association of type of oral anticoagulation with risk of bleeding in 45,114 patients with venous thromboembolism during initial and extended treatment-A nationwide register-based study. J Intern Med . Dec 2023;294(6):743-760. doi:10.1111/joim.13712. https://www.ncbi.nlm.nih.gov/pubmed/37641391
- Huma H, Rawat A, Kaur M, et al. Comparison of Effectiveness and Safety of Apixaban, Dabigatran, and Rivaroxaban in Patients With Valvular Atrial Fibrillation: A Network Meta-Analysis of Randomized-Control Trials and Observational Studies. Cureus. Apr 2024;16(4):e57656. doi:10.7759/cureus.57656. https://pubmed.ncbi.nlm.nih.gov/38707166/
- Ma F, Xu W, Chen J, Zhang J. Non-major bleeding risk of direct oral anticoagulants versus vitamin K antagonists for stroke prevention with atrial fibrillation: a systematic review and network meta-analysis. Eur J Clin Pharmacol . Aug 2023;79(8):1013-1022. doi:10.1007/s00228-023-03520-5. https://www.ncbi.nlm.nih.gov/pubmed/37310479
- Silverio A, Di Maio M, Prota C, et al. Safety and efficacy of non-vitamin K antagonist oral anticoagulants in elderly patients with atrial fibrillation: systematic review and meta-analysis of 22 studies and 440 281 patients. European heart journal Cardiovascular pharmacotherapy . Apr 9 2021;7(FI1):f20-f29. doi:10.1093/ehjcvp/pvz073. https://www.ncbi.nlm.nih.gov/pubmed/31830264
- Van Ganse E, Danchin N, Mahé I, et al. Comparative Safety and Effectiveness of Oral Anticoagulants in Nonvalvular Atrial Fibrillation: The NAXOS Study. Stroke. Jul 2020;51(7):2066-2075. doi:10.1161/strokeaha.120.028825. https://pubmed.ncbi.nlm.nih.gov/32539675/
- Talmor-Barkan Y, Yacovzada NS, Rossman H, et al. Head-to-head efficacy and safety of rivaroxaban, apixaban, and dabigatran in an observational nationwide targeted trial. European heart journal Cardiovascular pharmacotherapy . Dec 15 2022;9(1):26-37. doi:10.1093/ehjcvp/pvac063. https://pubmed.ncbi.nlm.nih.gov/36341531/
- Simon SJ, Patell R, Zwicker JI, Kazi DS, Hollenbeck BL. Venous Thromboembolism in Total Hip and Total Knee Arthroplasty. JAMA Netw Open . Dec 1 2023;6(12):e2345883. doi:10.1001/jamanetworkopen.2023.45883. https://www.ncbi.nlm.nih.gov/pubmed/38039005
- Yamashita Y, Morimoto T, Muraoka N, et al. Edoxaban for 12 Months Versus 3 Months in Patients With Cancer With Isolated Distal Deep Vein Thrombosis (ONCO DVT Study): An Open-Label, Multicenter, Randomized Clinical Trial. Circulation. Nov 21 2023;148(21):1665-1676. doi:10.1161/CIRCULATIONAHA.123.066360. https://www.ncbi.nlm.nih.gov/pubmed/37638968
- Gao F, Rahman F. DOACs and Atherosclerotic Cardiovascular Disease Management: Can We Find the Right Balance Between Efficacy and Harm. Curr Atheroscler Rep. Jun 2022;24(6):457-469. doi:10.1007/s11883-022-01022-w. https://www.ncbi.nlm.nih.gov/pubmed/35386093
- Al Aseri Z, AlGahtani FH, Bakheet MF, Al-Jedai AH, Almubrik S. Evidence-based Management of Major Bleeding in Patients Receiving Direct Oral Anticoagulants: An Updated Narrative Review on the Role of Specific Reversal Agents. J Cardiovasc Pharmacol Ther. Jan-Dec 2023;28:10742484231202655. doi:10.1177/10742484231202655. https://www.ncbi.nlm.nih.gov/pubmed/37872658
- Joosten LPT, van Doorn S, van de Ven PM, et al. Safety of Switching From a Vitamin K Antagonist to a Non-Vitamin K Antagonist Oral Anticoagulant in Frail Older Patients With Atrial Fibrillation: Results of the FRAIL-AF Randomized Controlled Trial. Circulation. Jan 23 2024;149(4):279-289. doi:10.1161/CIRCULATIONAHA.123.066485. https://www.ncbi.nlm.nih.gov/pubmed/37634130
- Rutherford OW, Jonasson C, Ghanima W, Soderdahl F, Halvorsen S. Effectiveness and safety of oral anticoagulants in elderly patients with atrial fibrillation. Heart. Mar 2022;108(5):345-352. doi:10.1136/heartjnl-2020-318753. https://www.ncbi.nlm.nih.gov/pubmed/33975877
- Suppah M, Kamal A, Saadoun R, et al. An Evidence-Based Approach to Anticoagulation Therapy Comparing Direct Oral Anticoagulants and Vitamin K Antagonists in Patients With Atrial Fibrillation and Bioprosthetic Valves: A Systematic Review, Meta-Analysis, and Network Meta-Analysis. The American journal of cardiology. Nov 1 2023;206:132-150. doi:10.1016/j.amjcard.2023.07.141. https://www.ncbi.nlm.nih.gov/pubmed/37703679
- Li D, Chang P, Zhang H, Bai F, Wu Q. The efficacy and safety of direct oral anticoagulants versus vitamin K antagonists in patients with left-sided bioprosthetic heart valves and atrial fibrillation: a systematic review and meta-analysis. Eur J Clin Pharmacol. Apr 2023;79(4):461-471. doi:10.1007/s00228-023-03463-x. https://www.ncbi.nlm.nih.gov/pubmed/36795127
- Martha JW, Pranata R, Raffaelo WM, Wibowo A, Akbar MR. Direct Acting Oral Anticoagulant vs. Warfarin in the Prevention of Thromboembolism in Patients With Non-valvular Atrial Fibrillation With Valvular Heart Disease-A Systematic Review and Meta-Analysis. Front Cardiovasc Med . 2021;8:764356. doi:10.3389/fcvm.2021.764356. https://www.ncbi.nlm.nih.gov/pubmed/35096994
- Su X, Yan B, Wang L, Lv J, Cheng H, Chen Y. Oral Anticoagulant Agents in Patients With Atrial Fibrillation and CKD: A Systematic Review and Pairwise Network Meta-analysis. American journal of kidney diseases : the official journal of the National Kidney Foundation . Nov 2021;78(5):678-689 e1. doi:10.1053/j.ajkd.2021.02.328. https://www.ncbi.nlm.nih.gov/pubmed/33872690
- Pomozi E, Nagy R, Fehervari P, et al. Direct Oral Anticoagulants as the First Choice of Anticoagulation for Patients with Peripheral Artery Disease to Prevent Adverse Vascular Events: A Systematic Review and Meta-Analysis. J Cardiovasc Dev Dis. Feb 3 2023;10(2)doi:10.3390/jcdd10020065. https://www.ncbi.nlm.nih.gov/pubmed/36826561
- Chen DY, Tseng CN, Hsieh MJ, et al. Comparison Between Non-vitamin K Antagonist Oral Anticoagulants and Low-Molecular-Weight Heparin in Asian Individuals With Cancer-Associated Venous Thromboembolism. JAMA Netw Open. Feb 1 2021;4(2):e2036304. doi:10.1001/jamanetworkopen.2020.36304. https://www.ncbi.nlm.nih.gov/pubmed/33533929
- Zhou H, Ye LL, Zhou JT, Ma FX, Ma JJ, Zhang JH. Direct oral anticoagulants (DOACs) versus low-molecular-weight heparin (LMWH) for extended thromboprophylaxis following major abdominal/pelvic cancer-related surgery: a systematic review and meta-analysis. Surgical endoscopy . Mar 2024;38(3):1131-1138. doi:10.1007/s00464-023-10649-y. https://www.ncbi.nlm.nih.gov/pubmed/38267639
- Zhou H, Chen TT, Ye LL, Ma JJ, Zhang JH. Efficacy and safety of direct oral anticoagulants versus low-molecular-weight heparin for thromboprophylaxis after cancer surgery: a systematic review and meta-analysis. World journal of surgical oncology. Feb 26 2024;22(1):69. doi:10.1186/s12957-024-03341-5. https://www.ncbi.nlm.nih.gov/pubmed/38403630
- Alhousani M, Malik SU, Abu-Hashyeh A, et al. Using oral anticoagulants among chronic kidney disease patients to prevent recurrent venous thromboembolism: A systematic review and meta-analysis. Thromb Res . Feb 2021;198:103-114. doi:10.1016/j.thromres.2020.11.036. https://www.ncbi.nlm.nih.gov/pubmed/33310644
- Schrag D, Uno H, Rosovsky R, et al. Direct Oral Anticoagulants vs Low-Molecular-Weight Heparin and Recurrent VTE in Patients With Cancer: A Randomized Clinical Trial. JAMA. Jun 13 2023;329(22):1924-1933. doi:10.1001/jama.2023.7843. https://www.ncbi.nlm.nih.gov/pubmed/37266947
- Coleman CI, Caroti KS, Abdelgawwad K, et al. Effectiveness and Safety of Rivaroxaban and Low Molecular Weight Heparin in Cancer-Associated Venous Thromboembolism. JACC CardioOncol. Apr 2023;5(2):189-200. doi:10.1016/j.jaccao.2022.10.014. https://www.ncbi.nlm.nih.gov/pubmed/37144109
- Bea S, Iyer GS, Kim DH, et al. Oral Anticoagulation and Risk of Adverse Clinical Outcomes in Venous Thromboembolism. JAMA Internal Medicine. 2025;doi:10.1001/jamainternmed.2025.1109. https://jamanetwork.com/journals/jamainternalmedicine/article-abstract/2833602
Wellness
Specialists
1-800-226-2370 - This service is FREE
7:30 AM - 12 AM (ET) Mon-Fri | 9 AM - 12 AM (ET) Sat-Sun